
Научная электронная библиотека
Монографии, изданные в издательстве Российской Академии Естествознания

Молекулярно-генетические исследования синдрома Ретта
Молекулярно-генетические исследования являются необходимым этапом диагностики заболевания. Эти исследования проводились у 354 детей с RTT. Мутации гена МЕСР2 у детей определяли методом прямого секвенирования. Анализ кодирующей последовательности гена MECP2 проведен в 350 спорадических случаях и у 2 пар конкордантных близнецов с RTT. Мутации гена MECP2 были обнаружены у 315 из 354 (89 %) детей. Большинство детей с мутациями в гене MECP2 были женского пола (352), за исключением двух мальчиков с классическим течением заболевания, у которых определен соматический мозаицизм по мутациям R168X и R270X. У мальчика с мутацией R270X наблюдался тканеспецифический мозаицизм по анеуплоидии хромосомы Х, обнаруженной в 14 % ядер мышечных клеток (кариотип – 47,XXY/46,ХY) (рис. 25). Несмотря на то, что случаи классического RTT у мальчиков с мутациями гена MECP2 являются редкими, в наших исследованиях наблюдалось два таких ребёнка. Опубликовано несколько десятков подобных случаев [Vorsanova et al., 1996; Villard et al., 2000; Hoffbuhr et al., 2001; Psoni et al., 2010]. У наблюдавшихся нами мальчиков обнаружен соматический мозаицизм по мутации гена MECP2, а у одного из них – тканеспецифический мозаицизм по синдрому Клайнфельтера (кариотип 47,XXY). Эти наблюдения подтверждают то, что мутации, приводящие к RTT у девочек, совместимы с жизнью у мальчиков лишь при наличии мозаичной или полной формы синдрома Клайнфельтера или при соматическом мозаицизме МЕСР2 мутаций [Vorsanova et al., 2001; Iourov et al., 2008 в, 2010]. У одной из пар конкордантных близнецов определена мутация R255X, у второй близнецовой пары выявлена мутация R270X. У 42 девочек с RTT мутаций в гене MECP2 обнаружено не было. Среди детей с классической формой RTT у большинства (249 из 262 больных, 95 %) определены MECP2-мутации. При атипичных формах заболевания доля больных с MECP2 мутациями была значительно ниже – 65 из 92 (70,7 %). Следует отметить, что у детей с врожденной формой RTT (2 ребенка) и формой с ранним началом судорог (9 детей) МЕСР2 мутаций не было обнаружено. Сравнение полученных нами данных с другими исследованиями свидетельствует о том, что частота мутаций гена МЕСР2 в группе наших детей была высокой: 95 % случаев классического и 70,7 % случаев атипичного синдрома. В зарубежных исследованиях показано, что частота мутаций в гене МЕСР2 при классической форме RTT варьирует от 63 до 100 %, а при атипичных формах заболевания – от 25 до 44 % [Cheadle et al., 2000; Monnerat et al., 2010]. Высокий процент мутаций гена MECP2 в наших исследованиях можно объяснить использованием шкалы количественной оценки фенотипа, а также эффективностью молекулярно-генетического метода, применявшегося в работе. На рис. 26 представлены примеры анализа мутаций гена МЕСР2 методом секвенирования ДНК.

Рис. 25. Интерфазная FISH с применением околоцентромерного ДНК зонда на хромосому Х, демонстрирующая наличие дополнительной
хромосомы Х у мальчика
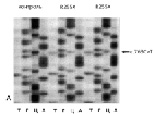
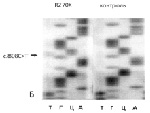
Рис. 26. Результаты анализа мутаций гена MECP2: А – мутация R255X (с.763C>T) у конкордантных близнецов (Т – тимин, Г – гуанин, Ц – цитозин, А – аденин). Б – мутация R270X (с.808С>Т) у мальчика с классической формой RTT. На электрофореграмме ребенка полоса невысокой интенсивности, соответствующая тимину в положении с.808, указывает на наличие мутации. Определяется полоса, соответствующая цитозину в том же положении, что указывает на присутствие аллеля без МЕСР2 мутации. Наличие двух аллелей свидетельствует о соматическом мозаицизме по мутации с.808С>Т у ребёнка
Миссенс мутации были обнаружены у 113 пациентов с RTT (36 %), нонсенс мутации – у 134 больных (44 %), мутации со сдвигом рамки считывания составили 60 случаев (19 %), 6 детей (2 %) имели делеции внутри рамки считывания (1067del276bp, 1085del132bp, 1156del42bp, 1157del45bp,1161del6bp, 1162del18bp). Рекуррентные мутации были определены в 201 из 315 случаев MECP2-мутаций (64 %), самой частой из них была мутация R255X (50 или 16 %). Частоты других рекуррентных мутаций составили 3 % для R106W, 7 % для R133C, 14 % для T158M, 11 % для R168X, 9 % для R270X, 3 % для R294X и 8 % для R306C. В целом частоты наблюдавшихся нами мутаций были близки частотам, приведенным в международной электронной базе данных по мутациям у больных с RTT [RettBASE: IRSF MECP2 Variation Database]. Некоторые различия были следствием немногочисленности исследованной выборки по сравнению с RettBASE, суммировавшей результаты большинства проведенных ранее работ. Наиболее частыми мутациями в нашем исследовании и в RettBASE являлись R255X, T158M, R168X, R270X, другие рекуррентные мутации R106W, R133C, R294X, R306C встречались несколько реже. В нашей работе у двух больных наблюдалась мутация G269fs (c.806delG), которая в RettBASE представлена 63 случаями. Возможно, что данную мутацию у детей с RTT следует отнести к разряду рекуррентных. У 5 девочек с атипичной формой RTT с ранним началом судорог при исследовании гена CDKL5 обнаружены патогенные варианты (с.463+1G>A, c.1153C>T, с.65dupG, c.2152delG, c.2419_2431del), что позволило установить у них атипичный вариант RTT, связанный с мутациями гена CDKL5. Мутации с.463+1G>A и с.65dupG были описаны ранее у детей с атипичным вариантом RTT [Nemos еt al., 2009; RettBASE]. Вариант c.1153C>T в 12 экзоне гена CDKL5 (chrX:18622197C>T) приводит к появлению сайта преждевременной терминации трансляции в 385 кодоне (p.Gln385X), данный вариант не зарегистрирован в контрольных выборках «1000 геномов», ESP6500 и ExAC. Поскольку мутация нарушает синтез полноразмерного белка со всех кодирующих транскриптов гена (согласно RefSeq), её следует расценивать как вероятно патогенную. Мутации c.2152delG, c.2419_2431del расценены как патогенные, поскольку ведут к сдвигу рамки считывания и связаны с фенотипом атипичного RTT (синоним – ранняя младенческая эпилептическая энцефалопатия, типа 2, OMIM 300672). Данные наблюдения подтверждают то, что часть случаев RTT связана с мутациями не только гена MECP2, но и других генов (CDKL5). Эти случаи могут служить подтверждением явления синдромального расщепления (syndrome splitting), когда клинически одно и то же заболевание оказывается результатом мутаций разных генов.
Поскольку из-за выраженного генетического полиморфизма молекулярно-генетическая диагностика Х-сцепленных форм умственной отсталости довольно трудоемка, и значительная часть случаев данной патологии остаётся недифференцированной, то особую значимость приобретает поиск общих для Х-сцепленных болезней биологических маркеров, которые можно использовать для их диагностики, а также для выявления женщин-носительниц Х-сцепленных мутаций. Одним из таких маркеров является неслучайная инактивация хромосомы Х.