
Научная электронная библиотека
Монографии, изданные в издательстве Российской Академии Естествознания

Белоцерковцева Л. Д., Коваленко Л. В., Корнеева Е. В., Майер Ю. И., Шишанок О. Ю., Ерченко Е. Н.,
ГЛАВА 2. ПАТОФИЗИОЛОГИЯ МЕТАБОЛИЧЕСКОГО СИНДРОМА
При изучении компонентов МС выделяют несколько уровней (табл.1). Основные пути метаболизма углеводов, жиров и белков тесно взаимосвязаны на уровне узловых метаболитов и ключевых ферментов [4;11].
Таблица 1. Уровни развития нарушений при метаболическом синдроме
Дискоординация метаболизма является первоосновой всех нарушений и основывается на существовании определенных ограничений во взаимных превращениях углеводов, жиров и белков, а именно: ограниченные обратные превращения из углеводов жиры, за счет глицерина; использование углеродного скелета, по крайней мере, 3/4 аминокислот (в том числе незаменимых) для глюконеогенеза и вовлечение углеводных структур в биосинтез лишь заменимых аминокислот; способности аминокислот свой углеродный скелет превращать частично или полностью в ацетил-КоА и, таким образом, служить материалом для синтеза жирных кислот. Эти ограничения усугубляются при инсулиновой недостаточности за счет изменения активности ряда ключевых ферментов обмена веществ, катализирующих фосфорилирова-ние глюкозы и фруктозо-6-фосфата, синтез гликогена из УДФ-1-глюкозы, фосфоролиз гликогена до глюкозо-1-фосфата, дефосфо-рилирование глюкозо-6-фосфата путем гидролиза до свободной глюкозы, превращение аминокислот в α-кетокислоты с помощью реакций переаминирования и окислительного дезаминирования, обратное превращение пировиноградной кислоты в фосфоенол-пируват, липолиз триглицеридов, образование ацетоновых тел из ацетил-КоА.
Помимо регуляторов, вмешивающихся в метаболические процессы на уровне ферментных реакций, существует влияние гормонов, связанное с их выбросом в кровеносное русло. Так, адреналин и норадреналин увеличивают скорость липолиза в жировой ткани за счет стимуляции аденилатциклазы адипоцитов и синтеза цАМФ. Действие глюкагона сходно с действием кате-холаминов. Инсулин оказывает противоположное адреналину и глюкагону действие на липолиз и мобилизацию жирных кислот. СТГ, АКТГ также оказывают стимулирующее влияние на липолиз, увеличивая содержание жирных кислот в плазме крови [11].
Жировая ткань обладает ауто-, пара- и эндокринной функцией и секретирует «адипоцитокины», обладающие различным биологическим действием, которые могут (при их избытке - ожирении), вызывать развитие сопутствующих ожирению осложнений, в том числе ИР: лептин, фактор некроза опухоли-а (TNF-а), ингибитор-1 активатора плазминогена (PAI), протеин, стимулирующий ацилирование (ASK), интерлейкин-6, интер-лейкин-8, ангиотензин-П, резистин, адипонектин, адипсин, протеин agouti, трансформирующий фактор роста-β, адипофилин [4; 11;41]. Многие исследователи рассматривают TNF-a, как медиатор ИР при ожирении. TNF-a снижает активность тирозинкиназы инсулинового рецептора, тормозит экспрессию внутриклеточных переносчиков глюкозы ГЛЮТ-4 в мышечной и жировой ткани. Как показано in vivo, TNF-a может действовать в синергизме с интерлейкинами-1 и 6, а также стимулировать секрецию лептина [33].
Лептин - «голос» жировой ткани, регулирует пищевое поведение, воздействуя на центр насыщения в гипоталамусе. К физиологическим эффектам лептина относятся: повышение тонуса симпатической нервной системы, усиление термогенеза в адипоцитах, снижение синтеза инсулина, снижение транспорта глюкозы, воздействуя на инсулиновый рецептор клетки. Выявлено стимулирующее действие лептина на секрецию гонадотро-пинов. В препубертатном периоде уровень лептина параллельно повышается с увеличением массы до максимальных значений с началом полового созревания. В пубертатном периоде повышается чувствительность к лептину. Ожирение может быть связано с дефицитом лептина и лептинорезистентностью [41]. Рецепторы лептина присутствуют и в яичниках, причем непосредственное влияние на стероидогенез в яичниках может быть как стимулирующим, так и ингибирующим (в эксперементах на животных есть данные о снижении инсулинзависимого синтеза прогестерона и Е2 в клетках гранулезы). Выявлено, что в течение менструального цикла уровень лептина постепенно нарастает на протяжении фолликулиновой фазы, достигая пика в лютеиновую фазу [10;11; 19; 27; 47].
Количество инсулина и лептина в циркуляторном русле прямо пропорционально массе жировых отложений, и их называют «сигналами ожирения» Повышенный уровень лептина при лептинорезистентности и МС обусловливает развитие гормональной дисфункции и висцерального ожирения. Глюкокор-тикоидная нестабильность (внутриклеточный гиперкортицизм) при метаболическом синдроме и инсулинорезистентности так же приводит к развитию висцерального ожирения [51].
В транспорте половых стероидов активную роль играет циркулирующий ГСПС (глобулин, связывающий половые стероиды, или тестостерон-эстрадиол связывающий глобулин). Установлено, что количество ГСПС определяется как наследственными факторами, так и наличием некоторой экстрагенитальной и генитальной патологии. Наличие положительной корреляции между уровнем ГСПС и ХС ЛПВП и обратной между ГСПС и ХС ЛПНП и ЛПОНП обуславливает наследование данного глобулина как генетического фактора риска развития атеросклероза сосудов головного мозга, ИБС и АГ [19]. Установлена прямая корреляция между содержанием эстрона, 17β-эстрадиола и индексом массы тела, обратная корреляция существует между последним показателем и уровнем ГСПС в сыворотке крови, что особенно характерно для пациенток в периоде постменопаузы [38]. Снижение уровня ГСПС в постменопаузе приводит к росту концентрации свободного тестостерона, относительной гиперандрогении и вносит определенный вклад в формирование абдоминального ожирения. Отмечено, что у женщин с гиноидным типом распределения жировой клетчатки уровень ГСПС выше, чем у таковых с андро-идным типом [48].
При исследовании сыворотки крови пациенток, страдающих сахарным диабетом, было установлено, что нарастание уровня глюкозы и повышение уровня инсулина ведут к увеличению свободного тестостерона и понижению уровня ГСПС [11]. Есть данные о том, что у женщин с анамнестически ранним менархе (до 13 лет), в репродуктивном возрасте уровень ГСПС в сыворотке крови более низкий по сравнению с женщинами с поздним менархе [2].
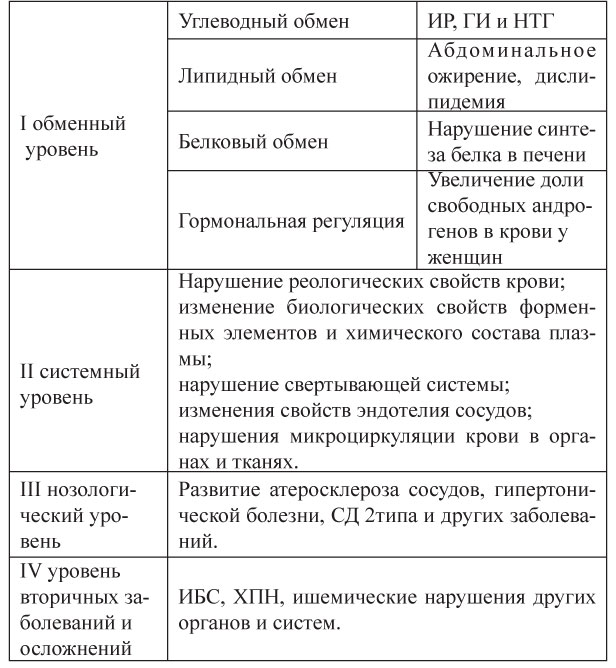
Ключевым моментом в первичных метаболических нарушениях является формирование инсулинорезистентности. Под ИР в настоящее время понимают первичное, селективное и специфическое нарушение биологического действия инсулина, сопровождающееся снижением потребления глюкозы тканями (преимущественно скелетными мышцами) и приводящее к хронической ГИ. Селективный характер ИР означает, что отдельные эффекты инсулина сохраняются, например, реабсорбция натрия в почечных канальцах или влияние на симпатический отдел нервной системы (схема 1).
Практически все составляющие МС являются факторами риска развития сердечно-сосудистых заболеваний, а в сочетании многократно ускоряют их развитие. Причем сочетания отдельных компонентов могут рассматриваться в рамках МС только при наличии ИР. Разумеется, не все компоненты метаболического синдрома встречаются одновременно. Каким фенотипом проявится метаболический синдром, зависит от взаимодействия факторов генетических и внешней среды.
Патогенез дислипидемии при метаболическом синдроме
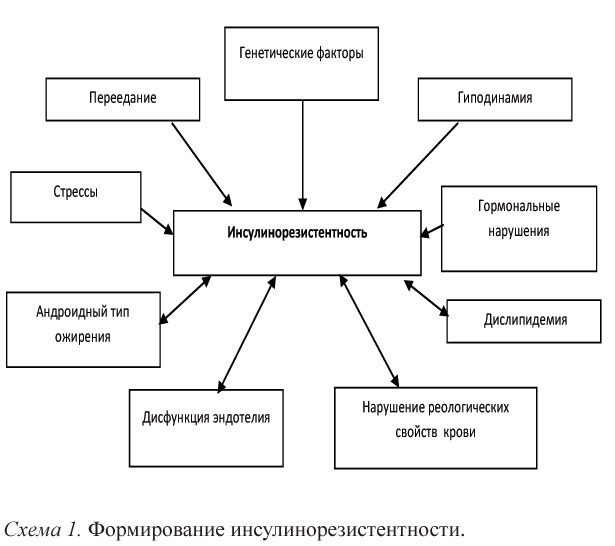
Наиболее частым вариантом дислипидемии при метаболическом синдроме является липидная триада: сочетание гипер-триглицеридемии, низкого уровня ХС ЛПВП и повышения фракции мелких плотных частиц ХС ЛПОНП, переносчиков тригли-церидов, что является результатом их повышенной печеночной продукции и сниженной элиминации. Механизм влияния ИР на развитие липидных нарушений представлены на схеме 2.
Гиперинсулинемия способствует увеличению пролиферации гладкомышечных клеток и фибропластов, увеличению активности рецепторов ХС ЛПНП и синтезу эндогенного ХС в клетках сосудистой стенки, коллагена, стимуляции выработки ИПФР.
Патогенез развития артериальной гипертензии при метаболическом синдроме
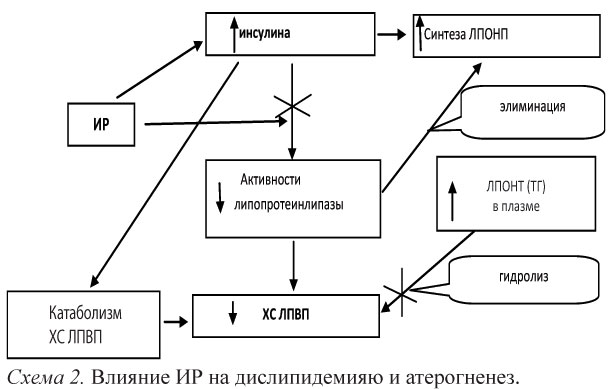
Механизм развития АГ при МС многогранен и неоднозначен. Общее влияние инсулина на АД представляет собой равновесие между прямым вазодилататорным и непрямым вазокон-стрикторным эффектами. При хронической ГИ возникает парадоксальная реакция со стороны сосудов в связи с преобладанием митогенного и симпатикостимулирующего компонентов (схема 3).
С другой сторон, возможен механизм, посредством которого АГ может сама способствовать ИР. Увеличение активности симпатической нервной системы, возникающей при АГ, вызывает понижение объемного кровотока в капиллярах скелетной мускулатуры в результате их вазоконстрикции, что увеличение пути диффузии глюкозы к клеткам и приводит к инсулинорезистент-ности.
Эндотелий сосудов обладает метаболической и секреторной активностью и играет ключевую роль в регуляции тонуса и проницаемости сосудов. Уникальное положение клеток эндотелия на границе между циркулирующей кровью и тканями делает их наиболее уязвимыми для различных патогенных факторов, находящихся в системном и тканевом кровотоке.
В настоящее время есть две основные точки зрения относительно формирования эндотелиопатии. Первая, что при синдроме ИР развивается дисфункция эндотелия сосудов и, в частности, нарушается синтез оксида азота в сосудистой стенке (оксид азота является мощным вазодилататором). Он оказывает сдерживающее влияние на пролиферацию гладкомышечных клеток, тормозит адгезию моноцитов к эндотелию сосудистой стенки, снижает перекисное окисление липидов, т.е. предохраняет стенки сосудов от повреждения. Существует также мнение, что дисфункция эндотелия является не следствием, а причиной в развитии ИР, одним из первичных дефектов, лежащих в основе ее развития. В случае первичного дефекта эндотелиальных клеток трансэндотелиальный транспорт инсулина нарушается, что может способствовать развитию ИР. Однако, до настоящего времени не получено достаточно данных в пользу первичной или вторичной роли эндотелиопатии в генезе инсулино-резистентности [4; 6]. Итак, основные механизмы повышения АД:
- объемозависимый
- повышение активности ренин-ангиотензин-адьдостеро-новой и симпатоадреналовой систем
- дисфункция эндотелия
- гиперлептинемия
- блокада вазодилатирующего эффекта инсулина.
Имеются данные, указывающие на снижение фибриноли-тической активности у пациентов с МС. PAI-1 является основнымингибитором активатора плазминогена, обеспечивая до 60% общейингибиторной активности в отношении активаторов плазминогена вплазме. Повышение уровня PAI-1 связано с риском тромбозов. PAI-1 синтезируется эндотелиальными клетками, моноцитами, макрофагами, гладкомышечными клетками, адипоцитами висцеральнойжировой ткани. Эндотелиальные клетки и тромбоциты регулируютвыделение PAI-1 в процессе фибринолиза. Было установлено, чтоГИ, гипергликемия и гипертриглицеридемия приводят к значительному повышению экспрессии гена, ответственного за продукциюPAI-1, и, соответственно, к повышению концентрации в крови. Внастоящее время считается, что повышение уровня PAI-1 являетсямаркером высокого риска инфаркта миокарда, ассоциируется ссахарным диабетом [4].