
Научная электронная библиотека
Монографии, изданные в издательстве Российской Академии Естествознания

Глава 3. Физические основы катализа реакции переэтерификации соединениями металлов
В настоящем разделе будут вкратце рассмотрены закономерности катализа модельной реакции переэтерификации соединениями металлов на примере традиционного катализатора тетрабутоксититана (ТБТ), как в случае использования только ТБТ, так и его совместного применения с нанонаполнителем (слюдой). Авторы [1] выполнили сравнительный кинетический анализ реакции переэтерификации без катализатора и в присутствии ТБТ с содержанием последнего 0,10 мольных % в расчете на реагент, взятый в недостатке. Этот анализ выполнен в рамках современных физических концепций [2-4].
Кинетические кривые степень конверсии-продолжительность реакции Q-t для реакции переэтерификации без катализатора и в присутствии ТБТ показаны на рис. 3.1. Обращает на себя внимание как количественное, так и качественное различие приведенных на рис. 3.1 кривых Q(t). Количественное различие выражается в гораздо более быстром росте Q по мере увеличения t, обусловленном наличием катализатора, что и следовало ожидать. Качественное различие отражается в изменении формы кривой Q(t): если в отсутствии ТБТ (некатализируемой реакции переэтерификации) получена линейная зависимость, которая указывает на протекание реакции в евклидовом (однородном) пространстве [5], то в присутствии ТБТ получена типичная криволинейная зависимость Q(t) c уменьшением скорости реакции dQ/dt по мере роста t. Такие реакции типичны для неоднородных (фрактальных) сред [8], вследствие чего они названы фракталоподобными. Зависимость dQ/dt от t в этом случае описывается соотношением (2.15), в котором показатель гетерогенности среды h варьируется в пределах 0<h≤1 для фрактальных сред и равен нулю для гамогенной (евклидовой) среды [6]. В последнем случае dQ/dt=const и зависимость Q(t) линейна, что и наблюдается для реакции переэтерификации без катализатора (рис. 3.1).
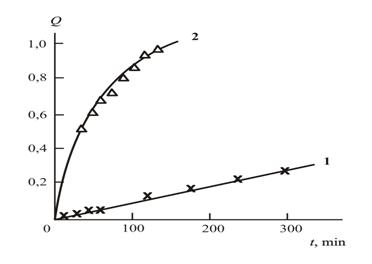
Рис. 3.1. Кинетические кривые степень конверсии–продолжительность реакции Q-t для реакции переэтерификации без катализатора (1) и в присутствии ТБТ(2)[1].
Рассмотрим причины фракталоподобной реакции переэтерификации в присутствии ТБТ. Эту реакцию, в общем, виде можно описать как реакцию рекомбинации реагентов А и В [4]:
А+В→ инертный продукт (3.1)
В рамках теории среднего поля спад концентрации реагента АρА при условии ρА= ρВ дается уравнением [4]:
![]() , (3.2)
, (3.2)
где k1-константа скорости первого порядка.
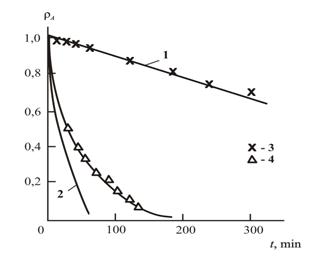
Рис.3.2. Зависимости спада концентрации реагента АρА от продолжительности реакции t для реакции переэтерификации без катализатора (1,3) и в присутствии ТБТ(2,4). 1,2–расчет по соотношению (3.2); 3,4– экспериментальные данные[1].
На рис. 3.2 приведено сравнение рассчитанных по соотношению (3.2) и полученных экспериментально функций ρА(t), где ρА определена как (1-Q). Как можно видеть, для реакции переэтерификации без катализатора спад ρА по мере роста t превосходно описывается в рамках теории среднего поля, тогда как в присутствии ТБТ спад ρА гораздо более медленный, чем предсказывает соотношение (3.2). Как известно [4], последний эффект обусловлен локальными флуктуациями распределения реагентов и в этом случае функция ρА(t) дается следующим образом:
ρА(t) ~ t-α , (3.3)
и
ρА(t) ~ ехр(-At-α), (3.4)
для малых и больших времен реакции, соответственно. В соотношениях (3.3) и (3.4) показатель α определяется размерностью пространства d, в котором протекает реакция, А- константа.
Для реакции рекомбинации описываемой уравнением (3.1), можно записать [4]:
![]() . (3.5)
. (3.5)
Отметим три важных аспекта, вытекающих из применения модели [4] для описания реакции переэтерификации. Во-первых, поскольку реакция переэтерификации с ТБТ и в его отсутствие протекают в идентичных условиях, то из сравнения кинетических кривых рис. 3.1 следует, что фракталоподобное поведение реакции в присутствии ТБТ обусловлено локальными флуктуациями распределения катализатора в реакционной среде. Во-вторых, деление продолжительности реакции на малые и большие времена связана с диффузией реагентов и катализатора [4]. При наличии локальных флуктуаций существуют достаточно малые области размером ξ, где положительные или отрицательные (т.е., избыток или недостаток) флуктуации ТБТ достаточно велики. Если мы получим характеристический масштаб времени tξ, которое требуется для прохождения диффундирующей частицей расстояния ξ, то условие t< tξ дает малые времена, а t>tξ – большие. И, в третьих, уравнение (3.5) дает величину показателя α для свободной диффузии реагентов. В случае принудительной (с преимущественной ориентацией) диффузии величина α определяется следующим образом [4]:
![]() , (3.6)
, (3.6)
т.е., для трехмерного пространства в случае принудительной диффузии α=1. В этом случае скейлинговый подход [4] дает соотношение (3.3), аналогичное уравнению (3.2). Это соответствие подтверждается данными рис. 3.2. Таким образом, указанный результат позволяет сделать вывод, что интенсивное перемешивание реакционной среды приводит к принудительной диффузии реагентов (метилбензоата и гептанола-1), но диффузия ТБТ остается свободной.
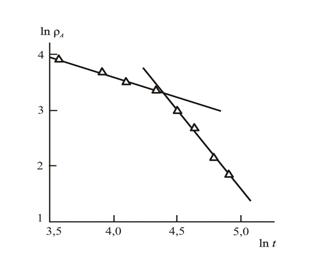
Рис.3.3. Зависимость спада концентрации реагента АρА от продолжительности реакции t в двойных логарифмических координатах для реакции переэтерификации в присутствии ТБТ[1].
На рис. 3.3 приведена зависимость ρА(t) в двойных логарифмических координатах, соответствующая соотношению (3.3), для реакции переэтерификации в присутствии ТБТ. Как можно видеть, эта зависимость распадается на два линейных участка с разными наклонами. Для первого участка (t<90 мин.) наклон равен ~0,75, т.е., соответствует уравнению (3.5) для протекания реакции в трехмерном евклидовом пространстве (d=3). Для второго участка (t>90 мин.) наклон равен ~3, т.е., не согласуется с возможными значениями этого показателя для реакции рекомбинации или других аналогичных реакций, для которых значение α ограничено сверху величиной 1,5 [2-4,7]. Это означает, что для рассматриваемой реакции переэтерификации времена меньше 90 мин. идентифицировать как малые, т.е., на этом временном интервале спад концентрации реагирующих частиц контролируется локальными флуктуациями распределения ТБТ, а времена, равные или больше 90 мин. - к большим, где ТБТ распределен равномерно. Это означает, что в последнем случае функция ρА(t) должна описываться соотношением (3.4). Действительно, приведенная на рис. 3.4 зависимость ρА от td/4 в логарифмических координатах является линейной, что подтверждает сделанное выше предположение.
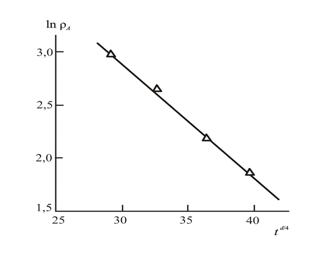
Рис.3.4. Зависимость спада концентрации реагента АρА от параметра td/4(d=3) в логарифмических координатах в случае больших времен для реакции переэтерификации в присутствии ТБТ[1].
Следовательно, реакция переэтерификации без катализатора может быть описана в приближении среднего поля, тогда как введение катализатора (ТБТ) определяет появление локальных флуктуаций ТБТ. Этот эффект приводит к фракталоподобной кинетике реакции переэтерификации. В этом случае реакция переэтерификации рассматривается как реакция рекомбинации и трактуется в рамках скейлинговых подходов. Практический аспект данного исследования очевиден – гомогенное распределение катализатора в реакционной среде или его принудительная диффузия позволяет уменьшить продолжительность реакции примерно в два раза [1].
Наиболее интересной особенностью реакции переэтерификации в присутствии 0,02 и 0,10 моль.% ТБТ является то, что увеличение концентрации катализатора в 5 раз практически не изменяет кинетику реакции и при малых временах величина Q для обеих концентраций ТБТ определяется одним общим уравнением [8]:
Q = 1-7,1t-d/4, (3.7)
где t дается в минутах, а d=3.
Следовательно, основным фактором, определяющим кинетику реакции переэтерификации в присутствии ТБТ, является не содержание катализатора, а его локальные флуктуации, увеличивающие продолжительность реакции в 2¸3 раза [8].
Как показано выше, введение 0,10 моль.% ТБТ резко ускоряет реакцию переэтерификации (константа скорости реакции k1 увеличивается более чем на порядок-от 0,18х10-4с-1 до 0,32 х10-3с-1), а также приводит к изменению типа кривой Q(t) от линейной к криволинейной (фракталоподобной, см. рис. 3.1). Расчет по соотношению (2.15) для реакции переэтерификации в присутствии ТБТ дал величину h=0,52. В свою очередь эффективная спектральная размерность ![]() реакционной среды определяется так [9]:
реакционной среды определяется так [9]:
![]() =2 (1-h). (3.8)
=2 (1-h). (3.8)
Таким образом, для реакции переэтерификации без катализатора (h=0, классический случай [6]) ![]() =2,0, а при введении ТБТ ее величина снижается до
=2,0, а при введении ТБТ ее величина снижается до ![]() =0,96 [10]. Это означает изменение степени связанности реакционной среды или появление в ней своего рода пустот, т.е., областей без катализатора. Величину
=0,96 [10]. Это означает изменение степени связанности реакционной среды или появление в ней своего рода пустот, т.е., областей без катализатора. Величину ![]() можно рассматривать как число взаимно ортогональных направлений на фрактальном множестве или дробное число степеней свободы [11]. Следовательно, введение ТБТ уменьшает указанные выше параметры примерно в два раза.
можно рассматривать как число взаимно ортогональных направлений на фрактальном множестве или дробное число степеней свободы [11]. Следовательно, введение ТБТ уменьшает указанные выше параметры примерно в два раза.
Количественное описание кинетики реакции переэтерификации можно получить с помощью следующего уравнения [12]:
Q=Аt![]() /2, (3.9)
/2, (3.9)
где А-константа, пропорциональная числу мест реакции.
Уравнение (3.9) хорошо описывает кривые Q(t) при следующих значениях его параметров: реакция переэтерификации без катализатора ![]() =2,0 и А=9,2х10-4 и для реакции в присутствии ТБТ
=2,0 и А=9,2х10-4 и для реакции в присутствии ТБТ ![]() =0,96 и А=9,2х10-2. Таким образом, введение ТБТ приводит к появлению существенной неоднородности (гетерогенности) реакционной среды (рост h) и соответствующему снижению степени ее эффективной связности, характеризуемой размерностью
=0,96 и А=9,2х10-2. Таким образом, введение ТБТ приводит к появлению существенной неоднородности (гетерогенности) реакционной среды (рост h) и соответствующему снижению степени ее эффективной связности, характеризуемой размерностью ![]() , а также к увеличению числа мест реакции, характеризуемого параметром А [10].
, а также к увеличению числа мест реакции, характеризуемого параметром А [10].
Следующим очевидным этапом этого раздела является исследование влияния нанонаполнителя (слюды) на кинетику реакции переэтерификации в присутствии катализатора (ТБТ), которое было выполнено в работе [13].
На рис.3.5 сплошными линиями показаны экспериментальные кинетические кривые степень конверсии – продолжительность реакции Q(t) для трех реакций переэтерификации с необработанной слюдой, с ТБТ и совместным присутствием слюды и ТБТ. Как можно видеть, скорость реакции возрастает именно в таком порядке (константы скорости реакции k1 равны 0,15х10-4, 3,15х10-4 и 3,90х10-4 с-1, соответственно). Если реакция переэтерификации без катализатора (ТБТ) имеет линейную кинетику Q(t) и, следовательно, моделируется в рамках теории среднего поля (см. уравнение (3.2)), то реакции в присутствии ТБТ имеют криволинейную кинетику, в которой спад концентрации реагентов более медленный по сравнению с линейным (см. рис. 3.2). Как показано выше, указанная криволинейность определяется гетерогенностью (фрактальностью) реакционной среды [6,9], обусловленной локальными флуктуациями распределения ТБТ [1,8,10].
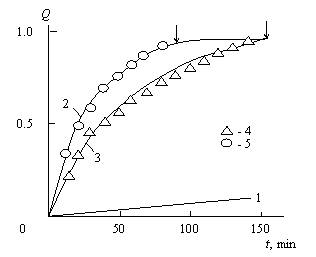
Рис.3.5. Экспериментальные (сплошные линии) и рассчитанные согласно уравнению (3.10) (точки) кинетические кривые Q(t) для реакции переэтерификации со слюдой (1), ТБТ (2, 4), слюдой и ТБТ (3, 5). [13].
На рис. 3.6 приведены зависимости ρА(t) в двойных логарифмических координатах, соответствующие соотношению (3.3). Как следует из этих графиков, при малых временах для реакций с ТБТ, как и выше, получены разные наклоны: для реакции без слюды α=0,75 или d/4, в присутствии слюды α=1,0 или (d+1)/4.

Рис.3.6. Зависимости ρА(t) в двойных логарифмических координатах, соответствующие соотношению (3.3), для реакции переэтерификации с ТБТ (1), со слюдой и ТБТ (2) [13].
Таким образом, интенсивное перемешивание реакционной среды вызывает принудительную диффузию ТБТ только в присутствии слюды. Этот эффект существенно ускоряет реакцию переэтерификации.
При больших временах реакция переэтерификации описывается соотношением (3.4) с α=0,75 или d/4 (см. рис. 3.4). Иначе говоря, в случае, когда большая часть ТБТ израсходована, перемешивание реакционной среды уже не может вызвать принудительную диффузию и обе реакции (со слюдой и без нее) описываются соотношением (3.4) с α= d/4, но с разными значениями коэффициента А.
Для объяснения перехода от свободной диффузии к принудительной в катализируемой реакции переэтерификации авторы [13] использовали фрактальную модель кинетики химических реакций [14,15]. Основное уравнение этой модели имеет вид [13]:
Q = Cohot(3-Df )/2 , (3.10)
Co - начальная концентрация реагентов, ho - исходная вязкость реакционной среды, Df - фрактальная размерность продукта реакции.
Отметим важный с точки зрения фракталов момент. Продукт модельной реакции переэтерификации (молекулу гептилбензоата) нельзя рассматривать как фрактальный объект в строгом физическом смысле этого термина, поскольку она, в отличие от макромолекулярного клубка [16], не обладает свойством самоподобия. Тем не менее, как отмечалось выше [17], в процессе переэтерификации кривая Q(t) имеет автозамедленный характер, а это означает, что молекула гептилбензоата ведет себя подобно фрактальному объекту и в силу указанных обстоятельств названа, следуя Копельману [6], фрактальноподобным объектом [18].
Из уравнения (3.10) следует, что построение зависимостей Q(t) в двойных логарифмических координатах в случае линейности этих зависимостей позволяет получить из их наклона величину Df фракталоподобной молекулы гептилбензоата. Такие зависимости приведены на рис. 3.7, они действительно линейны и по их наклону была определена величина Df=2,02, одинаковая в случае катализируемых ТБТ реакций переэтерификации как в присутствии слюды, так и без нее. Это означает, что разная форма кривых Q(t) для указанных реакций не является следствием разной структуры продукта реакции.
Достаточно очевидно, что введение слюды в реакционную смесь должно повысить ее вязкость η0. Теоретическую оценку степени такого повышения η0 можно выполнить в рамках модели Кернера, из которой для суспензий следует [19]:
![]() , (3.11)
, (3.11)
где ![]() и
и ![]() 0 – исходные вязкости реакционной среды в присутствии слюды и без нее, соответственноφн –объемная доля наполнител(слюды).
0 – исходные вязкости реакционной среды в присутствии слюды и без нее, соответственноφн –объемная доля наполнител(слюды).

Рис. 3.7. Зависимость Q(t) в двойных логарифмических координатах соответствующих уравнению (3.10), для реакции переэтерификации с ТБТ (1), слюдой и ТБТ (2) [13].
Если в качестве первого приближения принять равными объемную и массовые доли наполнителя, то уравнение (3.11) даст увеличение ![]() реакционной среды в присутствии слюды по сравнению с η0 в 2,07 раза.
реакционной среды в присутствии слюды по сравнению с η0 в 2,07 раза.
Далее, пологая в уравнении (3.10) величину с0=1,0 для реакционной среды без слюды и с0=0,7 для этой же среды в присутствии слюды, можно рассчитать величины η0 и ![]() в относительных единицах, которые равны 0,084 и 0,157, соответственно. Таким образом, эта оценка, дает
в относительных единицах, которые равны 0,084 и 0,157, соответственно. Таким образом, эта оценка, дает ![]() /
/![]() 0=1,88, что близко к полученной согласно уравнению (3.11). Следовательно, приведенные выше результаты показали, что переход от свободной диффузии в случае реакционной среды без наполнителя к принудительной диффузии в присутствии слюды обусловлен увеличением исходной вязкости
0=1,88, что близко к полученной согласно уравнению (3.11). Следовательно, приведенные выше результаты показали, что переход от свободной диффузии в случае реакционной среды без наполнителя к принудительной диффузии в присутствии слюды обусловлен увеличением исходной вязкости ![]() 0, что позволяет захват частиц ТБТ при перемешивании.
0, что позволяет захват частиц ТБТ при перемешивании.
Отметим еще один важный аспект. Для реакции переэтерификации в присутствии слюды без катализатора величина Df=1,71 [17,20]. Это означает, что молекула гептилбензоата «растягивается» на поверхности слюды [21], что приводит к снижению Df по сравнению с данными, полученными согласно рис. 3.7 (Df=2,02). Это наблюдение предполагает, что введение ТБТ переносит реакцию с поверхности слюды в объем реакционной смеси, одновременно внося флуктуации в процесс переэтерификации (сравните кривые 1, 2 и 3 рис. 3.5).
Кинетические кривые Q(t) можно моделировать с помощью уравнения (3.10). На рис. 3.5 приведены результаты такого моделирования при следующих параметрах: для катализируемой реакции без слюды с0=1,0, ![]() 0=0,084 и Df=2,02 и для этой же реакции переэтерификации в присутствии слюды с0=0,7 (с учетом занимаемого слюдой объема),
0=0,084 и Df=2,02 и для этой же реакции переэтерификации в присутствии слюды с0=0,7 (с учетом занимаемого слюдой объема), ![]() =0,157 и Df=2,02. Как можно видеть, кинетические кривые Q(t) в обоих указанных случаях хорошо моделируется в рамках фрактальной кинетики химических реакций. Далее можно теоретически оценить время завершения реакции переэтерификации tпр, полагая в уравнении (3.10) Q=1,0. На рис. 3.5 величины tпр (равные 90 и 145 мин. для реакций в присутствии слюды и без нее, соответственно) указаны стрелками. Из данных рис. 3.5 следует, что эта оценка также хорошо соответствует экспериментальным результатом.
=0,157 и Df=2,02. Как можно видеть, кинетические кривые Q(t) в обоих указанных случаях хорошо моделируется в рамках фрактальной кинетики химических реакций. Далее можно теоретически оценить время завершения реакции переэтерификации tпр, полагая в уравнении (3.10) Q=1,0. На рис. 3.5 величины tпр (равные 90 и 145 мин. для реакций в присутствии слюды и без нее, соответственно) указаны стрелками. Из данных рис. 3.5 следует, что эта оценка также хорошо соответствует экспериментальным результатом.
Следовательно, изложенные выше результаты предполагают, что увеличение скорости, катализируемой ТБТ реакции переэтерификации в присутствии нанонаполнителя (слюды) обусловлено переходом от свободной диффузии катализатора в отсутствии слюды к принудительной диффузии. Этот переход определяется увеличением исходной вязкости реакционной среды из-за присутствия нанонаполнителя. Указанный эффект позволяет снизить продолжительность реакции переэтерификации примерно в 1,6 раза [22].
И в заключение настоящей главы рассмотрим кинетику реакции переэтерификации в случае, когда ТБТ был нанесен на поверхность слюды, обработанной серной кислотой (СМК) [23]. На рис. 3.8 приведены кинетические кривые степень конверсии -продолжительность реакции Q(t) для всех пяти исследованных в настоящей главе типов реакции переэтерификации. Как отмечалось в главе 2, все эти реакции моделировались в рамках кинетики реакций первого порядка. Как следует из данных рис. 3.8, кинетика этих реакций очень различается, о чем можно судить по величине константы скорости реакции первого порядка k1: она варьируется от 0,18х10-4 до 29х10-4с-1. Наименьшее значение k1 имеет реакция переэтерификации без катализатора и нанонаполнителя, наибольшее - реакция в присутствии СМК, обработанной ТБТ. Таким образом, СМК действует как катализатор реакции переэтерификации (подробно см. главу 2), хотя и более слабый, чем ТБТ, а обработка слюды ТБТ дает синергический
эффект. Рассмотрим физические основы наблюдаемого эффекта. Как было показано выше, молекулу гептилбензоата можно рассматривать как фракталоподобный объект с размерностью Df. В этом случае для описания реакции переэтерификации может быть использована модель диффузионно-ограниченной агрегации (ДОА), модифицированная с учетом наличия в реакционной среде многих мест протекания реакции, т.е., активных центров с концентрацией сакт [24]. Согласно этой модели, величину сакт можно оценить с помощью соотношения [24]:
![]() , (3.12)
, (3.12)
где Rq-радиус инерции фракталоподобной молекулы гептилбензоата, с0- концентрация частиц, формирующих кластер (гептилбензоата), т.е., концентрация реагентов.
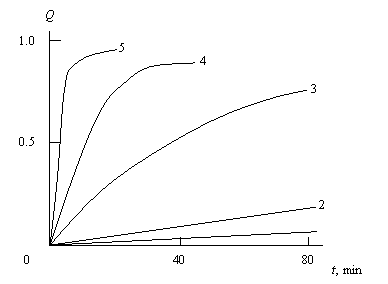
Рис.3.8. Кинетические кривые Q(t) для пяти типов реакции переэтерификации: 1-в отсутствии СМК и ТБТ; 2- в присутствии СМК, 3- в присутствии ТБТ; 4- в присутствии СМК и ТБТ; 5- в присутствии СМК обработанной ТБТ [23].
Поскольку гептилбензоат является низкомолекулярным веществом, для которого нет оснований ожидать роста молекулярной массы аналогично полимерам и, следовательно, увеличения Rq, то в работе [23] было принято Rq=сonst=10 относительных единиц. Во всех пяти исследуемых реакциях состав реагентов был одинаков и поэтому с0= сonst. Определить величину размерности Df можно с помощью уравнения (2.9), оценки согласно которому показали, что величина Df для исследуемых реакций снижается от 1,95 до 1,13, а величина сакт увеличивается от 1,1 до 7,4 относительных единиц. Другой способ описания реакции переэтерификации дает соотношение (2.10), в котором константа А пропорциональна числу захватов (объемов протекания реакции) [25].
Расчет по соотношению (2.10) показал, что величина А увеличивается по мере роста константы скорости реакции переэтерификации от 5 до 720 относительных единиц, т.е., она растет гораздо быстрее, чем сакт. Получить линейную корреляцию между А и сакт удалось только в форме кубической функции, т.е., А~ с3 акт (рис. 3.9). Такая зависимость предполагает, что захватом является объем реакционной среды с радиусом R, равным [24]:
![]() , (3.13)
, (3.13)
где d-размерность евклидова пространства, в котором протекает реакция, равная 3 в рассматриваемом случае, а согласно графику рис. 3.9 величина А может быть определена как R-1 [23].

Рис.3.9. Соотношение между числом захватов А и активных мест сакт для пяти исследуемых типов реакции переэтерификации [23].
На рис. 3.10 приведена зависимость k1 от сакт, которую вновь можно линеаризовать только в форме кубической функции. Исходя из сказанного выше, график рис. 3.10 предполагает, что реакция протекает в объеме, пропорциональном ![]() =
= ![]() и чем больше сакт, тем меньше этот объем, тем больше вероятность реакции (тем больше число захватов реагентов).
и чем больше сакт, тем меньше этот объем, тем больше вероятность реакции (тем больше число захватов реагентов).

Рис. 3.10. Зависимость константы скорости первого порядка k1 от числа активных мест сакт для пяти типов исследуемых реакций переэтерификации [23].
Следовательно, увеличение числа активных центров реакции приводит к снижению локального объема реакционной среды, в котором эта реакция может быть реализована. По своему физическому смыслу это означает ослабление пространственной флуктуации, что приводит к существенному росту скорости реакции переэтерификации. Очевидно, что с практической точки зрения наиболее выгодна реакция, катализируемая обработанной тетрабутоксититаном слюдой СМК[23].
И в заключение настоящей главы рассмотрим процесс катализа боридом молибдена Мо2В в реакции жидкофазного эпоксидирования этилаллилэтилакрилата (ЭАЭА) гидропероксидом трет-бутила (ГПТБ). В указанной реакции наблюдается нелинейная зависимость начальной скорости эпоксидирования Wо от содержания катализатора [кат.] [26]. Такой эффект был объяснен уменьшением степени редиспергирования агрегатов Мо2В по мере роста [кат.] в реакционной среде и соответствующим уменьшением доступной для катализа площади поверхности агрегатов Мо2В. Тем не менее, авторы [26] признали, что в рассматриваемом ими случае этот эффект не может полностью объяснить полученные экспериментальные данные.
Известно, что используемые в настоящее время катализаторы имеют шероховатую поверхность, которая, как правило, является фрактальной [27-29]. Кроме того, как показано в работе [30], дисперсные порошки обладают фрактальной поверхностью частиц, чья размерность dn изменяется в максимально широком диапазоне (2< dn £ 3). Эти обстоятельства позволяют предположить, что поверхность Мо2В также имеет фрактальную структуру.
Еще одним доводом в пользу такого предположения служит нелинейность зависимости Wo(кат), что является типичным признаком фрактальных реакций, т.е., либо реакций фрактальных объектов, либо реакций, протекающих во фрактальных пространствах [31,32]. Исходя из этого, авторы [33] продемонстрировали возможность количественного описания обнаруженных в работе [26] эффектов в рамках фрактального анализа.
Как известно [34], одним из наиболее примечательных свойств поверхности является ее способность контролировать физические и химические процессы относительно пути и скорости. Для диффузионно-контролируемых каталитических реакций в работе [34] получено следующее соотношение:
Q(t) ~ t (d-dn)/2, (3.14)
где Q - число частиц, прореагировавших на поверхности катализатора или степень конверсии, t - продолжительность реакции, d - размерность евклидова пространства, в котором протекает реакция (очевидно в рассматриваемом случае d = 3), dn - размерность поверхности катализатора.
Как показано в работе [34], соотношение (3.14) является достаточно общим и может быть использовано для описания любой реакции контролируемой диффузией. Так, авторы [14] использовали это соотношение для описания реакции полимеризации в форме уравнения (3.10).
Дифференцируя уравнение (3.14) по времени и используя аналогию с уравнением (3.10), можно получить [33]:
Wo ([кат], t) ~ [кат] t (1-dn)/2, (3.15)
поскольку при применении [кат] в силу его малой абсолютной величины сколько-нибудь значительного изменения hо не ожидается.
Чтобы рассчитать величину Wo согласно соотношению (3.15), необходимо оценить фрактальную размерность dn поверхности агрегатов исходных частиц Мо2В независимым способом. Для этой цели было использовано соотношение [30,34]:
Su ~ R d n -3, (3.16)
где Su - удельная площадь поверхности исходного порошка Мо2В, равная 0,31м2/2 [26], R - радиус агрегатов частиц Мо2В, принятый по данным [26], в интервале 1,4 ¸2,4 мкм при вариации [кат] = 0,5¸6,0 г/дм3.
Полагая для [кат] = 0,5 г/дм3 и соответствующей ей величине R = 1,4мкм dn = 2,2 [27], получим константу пропорциональности в соотношении (3.16), что позволяет расчет dn как функции [кат]. Эта зависимость приведена на рис.3.11 (сплошная линия), откуда следует быстрый рост dn при небольших [кат] и выход на плато - для [кат]>4г/дм3. Такая форма зависимости dn от концентрации исходного дисперсного материала является типичной и наблюдается для других условий агрегации, в частности, агрегации порошка графита с размером исходных частиц ~10 мкм в полимерных композитах [35], которая описывалась следующим эмпирическим уравнением:
![]() , (3.17)
, (3.17)
где dо и Ro - фрактальная размерность поверхности и радиус частиц исходного порошка, соответственно.

Рис.3.11. Зависимость фрактальной размерности dn поверхности агрегатов частиц катализатора Мо2В от его содержания [кат], рассчитанные согласно соотношениям (3.16) (1), (3.17) (2) и (3,15) (3) [33].
На рис. 3.11 также приведен расчет dn согласно уравнению (3.17) при dn =2,08 и Ro = 1,4 мкм. Как можно видеть получено достаточно хорошее соответствие зависимостей dn ([кат]), рассчитанных по уравнению (3.16) и (3.17), хотя величины dо для них несколько различаются (dо =2,20 и 2,08), соответственно.
Далее можно теоретически рассчитать величину Wo, используя соотношение (3.15). На рис. 3.12 приведено сравнение экспериментальных и рассчитанных указанным образом зависимостей Wo([кат]), где величины dn были оценены согласно уравнениям (3.16) и (3.17). Как можно видеть при обоих способах оценкиdn фрактальная модель катализа [34] дает достаточно точное как качественное, так и количественное описание изменения Wo при вариации содержания катализатора Мо2В. Иначе говоря, модель [34] дает корректное объяснение описанному в работе [26] эффекту. Как следует из соотношения (3.15), приdn=2 (гладкая поверхность катализатора) зависимость Wo от [кат] будет линейной при t = const (кривая 1 на рис. 3.12), но Wo при [кат] = const является функцией времени, снижаясь по мере роста t. При dn = 1 зависимость Wo от [кат] также будет линейной и не будет зависеть от t [33].
Используя соотношение (3.15), можно выполнить и обратную процедуру - рассчитать dn по Wo, вновь полагая dn = 2,2 при [кат] = 0,5 г/дм3 . Сравнение зависимостей dn ([кат]), рассчитанных согласно соотношениям (3.15) и (3.16), показано на рис 3.11. Несмотря на определенные расхождения, и ход этих зависимостей, и абсолютные величины dn достаточно хорошо согласуются (расхождение меньше 10%). Это обстоятельство указывает, что на начальной стадии катализа "отравления" катализатора не происходит [34]. Если бы такой эффект имел место, то полученные согласно соотношению (3.15) значения dn были бы меньше соответствующих величин, рассчитанных из соотношения (3.16) [34].
Далее рассмотрим природу частиц катализатора. Как известно [26], плотность r этих агрегатов можно определить из соотношения:
![]() . (3.18)
. (3.18)
Расчет r по уравнению (3.18) при Sn. = 0,31 м2/г = const и R = 1,4¸2,4 мкм показал снижение r по мере роста R в пределах 6910¸4030 кг/м3. Как известно [26], плотность частиц исходного катализатора Мо2В rо равна 9260 кг/м3. Таким образом, для агрегатов частиц выполняется условие r < rо, что типично для рыхлых фрактальных агрегатов плотность которых определяется уравнением (1.33). Расчет согласно этому уравнению показал, что фрактальная размерность агрегатов частиц Мо2В df в интервале R =1,4¸2,4 мкм изменяется незначительно, а именно в пределах df =2,13¸2,05. Отметим, что такая величина df типична для агрегатов, перестраивающихся во время процесса роста, т.е., включающих процессы реструктуризации [36].

Рис. 3.12. Зависимость начальной скорости эпоксидирования Wo от содержания катализатора [кат]: экспериментальные данные (1), расчет согласно соотношению (3.15) при dn, определенной по уравнениям (3.16) (2) и (3.17) (3) и при dn = 2 (4). ([ЭАЭА]о = 2,2 моль/л, [ГПТБ]о = 0,52 моль/л), Т=383К[26])[33].
Это указывает, что увеличение dn по мере роста [кат] (см. рис 3.11) обусловлено только процессом агрегации исходных частиц катализатора. Эта величина df также хорошо согласуется с размерностью агрегатов частиц никеля, полученной экспериментально [37].
И, наконец, выясним физический смысл структурного фактора катализатора, контролирующего скорость эпоксидирования. Процесс катализа можно рассматривать как процесс роста фрактального объекта с той разницей, что вместо частиц, присоединяющихся к фрактальному объекту, мы будем учитывать реагирующие молекулы. Согласно [38], полагаем, что агрегат частиц катализатора радиусом Rимеет три типа мест:
1) уже посещенные молекулами ("зараженные" или "мертвые") места, принадлежащие кластеру;
2) уже посещенные, но заблокированные ("невосприимчивые") места;
3) еще не посещенные места, являющиеся соседями посещенных (места роста).
Узлы роста образуют "открытую границу" фрактала [39] и их множество Ng имеет фрактальную размерность dg:
Ng ~ R dg . (3.19)
Между массой кластера (агрегата) N и Ng существует следующее соотношение [38]:
Ng ~ Nх, (3.20)
где х = dg / df= 1/2. Поскольку N ~ R df, то из соотношений (3.19) и (3.20) получим [33]:
Ng ~ N dg/df ~ R df df/df ~R dg ~ R df/2. (3.21)
Как показано выше, df »2 и тогда Wo ~ Ng ~ R. Этот вывод подтверждается данными рис. 3.13, где приведена зависимость Wo( R).
Следовательно, изложенные выше результаты показали, что методы фрактального анализа качественно и количественно объясняют полученный авторами [26] нелинейный характер зависимости начальной скорости эпоксидирования от содержания катализатора. Отметим, что континуальные модели, использующие представления евклидовой геометрии, не позволяют получить адекватного объяснения.
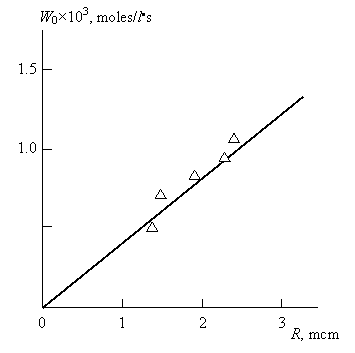
Рис.3.13. Зависимость начальной скорости эпоксидирования Wo от радиуса агрегатов частиц катализатора Мо2В R [33].
Литература к Главе 3.
1. Naphadzokova L. Kh., Kozlov G.V., Zaikov G.E. In book: Monomer and Polymer Research Frontiers. Ed. D’Amore A., Zaikov G.E. New York, Nova Science Publishers, Inc., 2006, p. 69-75.
2. Grassberger P., Procaccia I. J.Chem. Phys., 1982, v. 77, № 12, p. 6281-6284.
3. Meakin P., Stanley H.E. J. Phys. A, 1984, v. 17, № 1, p. L173-L177.
4. Kang K., Redner S. Phys. Rev. Lett., 1984, v. 52, № 12, p. 955-958.
5. Козлов Г.В., Заиков Г.Е. Теоретические основы химической технологии, 2003, т. 37, № 5, с. 555-557.
6. Копельман Р.В. В кн.: Фракталы в физике. Ред. Пьетронеро Л., Тозатти Э. М., Мир, 1988, с. 524-527.
7. Redner S., Kang K. J. Phys. A, 1984, v. 17, № 2, p. L451-L459.
8. Нафадзокова Л.Х., Овчаренко Е.Н., Козлов Г.В. Фундаментальные исследования, 2006, № 4, с. 101-102.
9. 9.Klymko P.W., Kopelman R. J. Phys. Chem., 1983, v. 87, № 23, p. 4565-4567.
10. Нафадзокова Л.Х., Гринева Л.Г., Козлов Г.В. Обозрение прикладной и промышленной математики, 2006, т. 13, № 1, с. 126-127.
11. Зеленый Л.М., Милованов А.В. Успехи физических наук, 2004, т. 174, № 8, с. 809-852.
12. Kozlov G.V., Zaikov G.E. The Structural Stabilization of Polymers: Fractal Models. Leiden-Boston, Brill Academic Publishers, 2006, 345 p.
13. Нафадзокова Л.Х., Козлов Г.В., Заиков Г.Е. Конструкции из композиционных материалов, 2008, №1, с.71-75.
14. Новиков В.У., Козлов Г.В., Успехи химии, 2000, т. 69, № 4, с. 378-399.
15. Kozlov G.V., Shustov G.B., Zaikov G.E. J. Balkan Tribologic. Assoc., 2003, v. 9, № 4, p. 467-514.
16. Havlin S., Ben-Avraham D. Phys. Rev. A, 1982, v. 26, № 3, p. 1728-1734.
17. Нафадзокова Л.Х., Карданов Х.М., Козлов Г.В. В сборн. научн. трудов «Седьмой регион. Наука и практика». Нальчик, Полиграфсервис т Т, 2005, с. 84-90.
18. Козлов Г.В., Нафадзокова Л.Х. Известия ВУЗов, Северо-Кавказск. регион, естествен. науки, 2006, № 3, с. 36-39.
19. Mills N.J. J.Appl. Polymer Sci., 1971, v. 15, № 11, p. 2791-2805.
20. Нафадзокова Л.Х., Козлов Г.В., Тленкопачев М.А. В сборн. статей VI Междунар. междисциплинарн. симпозиума «Фракталы и прикладная синергетика, ФиПС-2005». М., Интерконтакт Наука, 2005, с. 115-118.
21. Пфейфер П. В кн.: Фракталы в физике. Ред. Пьетронеро Л., Тозатти Э. М., Мир, 1988, с. 72-81.
22. Нафадзокова Л.Х., Козлов Г.В., Шустов Г.Б. Матер. междунар. конф. «Органическая химия от Бутлерова и Бейльштейна до современности». СПб., 26-29 июня, 2006, с. 614-615.
23. Буря А.И., Нафадзокова Л.Х., Козлов Г.В., Жукова А.И. Вопросы химии и химической технологии, 2007, №2, с.73-75.
24. Witten T.A., Meakin P. Phys. Rev. B, 1983, v. 28, № 10, p. 5632-5642.
25. Джорджевич З. В кн.: Фракталы в физике. Ред. Пьетронеро Л., Тозатти Э. М., Мир, 1988, с. 581-585.
26. Комаренская З.М., Никипанчук М.В., Трач Ю.Б., Яремко З.М., Федушинская Л.Б. Вопросы химии и химической технологии, 2002, № 1, с. 38-41.
27. Farin D., Peleg S., Yavin D. Langmuir, 1985, v. 1, № 4, p. 399-407.
28. Meakin P. Chem. Phys. Lett., 1986, v.123, 1983, v. 28, № 5, p. 428-432.
29. Wellner E., Pojanski D., Ottolenghi M., Huppert D., Avnir D. J. Amer. Chem. Soc., 1987, v. 109, № 3, p. 575-578.
30. Avnir D., Farin D., Pfeifer P. Nature, 1984, v. 308, № 5959, p. 261-263.
31. Kozlov G.V., Zaikov G.E. J. Balkan Tribologic Assoc., 2004, v. 10, № 1, p. 1-30.
32. Kozlov G.V., Zaikov G.E. In book: Handbook of Polymer Research, v. 20, Ed. Pethrick A., Zaikov G. New York, Nova Science Publichers, Inc., 2006, p. 85-117.
33. Kozlov G.V., Afaunova Z.I., Zaikov G.E. In book: Theoretical and Practical Guide to Organic Physical Chemistry. Ed. Zaikov G., Kozlov G., Makitra R. New York, Nova Science Publichers, Inc., 2006, p. 21-27.
34. Pfeifer P., Avnir D., Farin D. J. Stat. Phys., 1984, v. 36, № 5/6, p. 699-716.
35. Kozlov G.V., Lipatov Yu.S. Composite Interfaces, 2002, v. 9, № 6, p. 509-527.
36. Kolb M. J. Phys. A, 1986, v. 19, № 5, p. L263-L268.
37. Шамурина М.В., Ролдугин В.И., Прямова Т.Д., Высоцкий В.В. Коллоидный журнал, 1994, т. 56, № 3, с. 451-454.
38. Стенли Х. В кн.: Фракталы в физике. Ред. Пьетронеро Л., Тозатти Э. М., Мир, 1988, с. 463-477.
39. Rammal R., Toulouse G. J. Phys. Lett. (Paris), 1983, v. 44, № 1, p. L13-L22.