
Научная электронная библиотека
Монографии, изданные в издательстве Российской Академии Естествознания

2.1. Основные принципы фрактальной модели процессов газопереноса
Диффузия на фрактальных объектах, называемая также аномальной [1] или странной [2], имеет ряд специфических особенностей, исследование которых начато более 20 лет тому назад [3] и продолжается до сих пор. Эти особенности полностью относятся и к полимерам, которые обладают фрактальной структурой. Кроме того, на указанные особенности налагается специфика структуры полимеров, в которых диффузия реализуется через микрополости флуктуационного свободного объема [4; 5]. Эти обстоятельства делают процессы диффузии в полимерных мембранах достаточно сложными и зависящими от значительного числа параметров. Сначала мы рассмотрим трактовку процессов газопереноса в рамках кластерной модели. Поскольку весь флуктуационный свободный объем полимера концентрируется в рыхлоупакованной матрице [6], то следует ожидать, что повышение ее относительной доли jр.м. приведет к увеличению коэффициентов диффузии D или проницаемости Р. Действительно, такая корреляция получена в работе [7], которая аналитически выражается следующими простыми соотношениями [8]:
![]() , (2.2)
, (2.2)
где ![]() и
и ![]() - константы, fc - относительный флуктуационный свободный объем.
- константы, fc - относительный флуктуационный свободный объем.
Наличие скачка коэффициента диффузии D при температуре плавления Тпл полиэтиленов является хорошо установленным экспериментальным фактом [9]. Авторы [9] предложили теоретическую модель для описания температурной зависимости D, которая предполагает два разных уравнения для температурных интервалов T < Tпл и T > Tпл. Отметим, что эта модель не учитывает структурных различий аморфной фазы полиэтиленов в аморфно-кристаллическом (T < Tпл) и вязкотекучем (T > Tпл) состояниях. В работах [10-13] рассмотрено описание зависимости D(T) и, в частности, скачка D при Тпл, в рамках кластерной модели, которая учитывает указанные выше структурные различия.
Рассмотрим физические основы существования скачка D при Тпл в предлагаемой трактовке. Модель [9] предполагает одинаковую структуру аморфной фазы в аморфно-кристаллическом полиэтилене и его расплаве. В этом случае роль кристаллической фазы сводится к уменьшению D, пропорциональному степени кристалличности. Кроме того, авторы [9] предположили, что лимитирующей стадией диффузии газов в аморфно-кристаллических полиэтиленах является перенос через места контакта кристаллитов, что следует считать достаточно произвольным предположением. Известно [14], что переход кристаллической фазы в аморфную является наиболее плотноупакованной частью некристаллических областей полиэтиленов и поэтому нет оснований ожидать как непосредственного контакта кристаллитов, так и повышенной рыхлости в местах таких контактов, если они существуют [11].
В рамках предлагаемой трактовки структура аморфной фазы аморфно-кристаллического полиэтилена и его расплава различаются наличием в первой областей локального порядка (кластеров), через которые процессы газопереноса также не реализуются. Появление этих областей при Т £ Тпл обусловлено процессом кристаллизации и вызванным этим процессом натяжением цепей в аморфных прослойках [15]. При Т > Тпл также сохраняется некоторая доля областей локального порядка, но в этом случае он будет динамическим, с небольшим временем жизни [16]. Скачок D при Тпл в рамках самых общих физических представлений обусловлен спецификой изменения параметра порядка при критической температуре Ткр (для полиэтиленов Ткр = Тпл). На рис. 2.1 показана схематическая температурная зависимость параметра порядка j(Т), из которой следует очень быстрый рост j в узком интервале температур Т £ Тпл [17]. Как отмечалось в предыдущей главе, для описания зависимости jкл(Т) (параметра порядка) может быть использована концепция термического кластера, где величина jкл дается уравнением [18]:
![]() , (2.3)
, (2.3)
где индекс bТ = 0,40 [13].
Для принятого в работе [9] температурного интервала скачка D примерно 5К и Тпл = 403К [19], получим jкл = 0,176, что и является причиной указанного скачка [12].
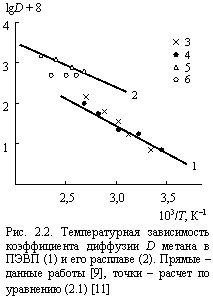
Сравнение температурных зависимостей коэффициента диффузии для ПЭВП и его расплава, рассчитанной по уравнению (2.1) и полученной экспериментально [9], приведено на рис. 2.2. Получено неожиданно хорошее (с учетом аппроксимационного характера уравнения (2.1) [8]) соответствие теории и эксперимента, а постоянная величина D при T > Tпл обусловлена принятым в [11] условием jр.м. = const = 0,933 в этом температурном интервале. Важно также отметить, что использована одна и та же константа , равная 3,65 ´ 10-11 м2/с для обоих температурных интервалов [10].
Следовательно, кластерная модель позволяет объяснить поведение коэффициента диффузии как функции температуры в рамках единого структурного подхода как выше, так и ниже Тпл. Предложенная авторами [10-13] трактовка объясняет скачок D при Тпл как результат формирования (распада) областей локального порядка (кластеров), непроницаемых для газопереноса. Рассматриваемая трактовка дает не только качественное, но и количественное описание зависимости D(Т) во всем использованном интервале температур.
Авторы [22] предложили фрактальную модель диффузии газов на примере аморфно-кристаллического полиэтилена, учитывая его более сложную структуру по сравнению с аморфными полимерами. Присутствие кристаллитов в диффузионной среде снижает суммарную скорость переноса через равные объемы. Чем выше степень кристаллитов капилляров или пустот, тем ниже коэффициенты диффузии и растворимости и, следовательно, проницаемость [4; 23]. Однако, обычно величина D не прямо пропорциональна содержанию аморфной фазы, как в случае растворимости [9], а носит более сложный характер [4].
В связи с этим были введены эмпирические коэффициенты, учитывающие два основных фактора, влияющих на диффузионные характеристики аморфно-кристаллических полимеров. Первый из них - коэффициент непрямолинейности или извилистости t - учитывает тот факт, что путь молекулы пенетранта в аморфной фазе отличается от прямолинейного, что снижает коэффициент диффузии D (t ³ 1). Второй коэффициент bм учитывает снижение скорости диффузии, вызванное ограничением подвижности цепей в аморфной фазе из-за наличия кристаллитов. Коэффициент bм называется коэффициентом неподвижности цепей [4]. С учетом этих фактором величина D аморфно-кристаллического полимера записывается так [4]:
![]() , (2.4)
, (2.4)
где Dа - коэффициент диффузии соответствующего полностью аморфного полимера; jа - относительная доля аморфной фазы.
Наличие двух эмпирических коэффициентов в уравнении (2.4) (t и bм) делает теоретическое предсказание D практически невозможным.
Кроме указанных факторов, на величину D существенное влияние оказывает размер молекулы dм пенетранта [4; 5; 23]. В общем случае увеличение dм приводит к очень резкому снижению D. Достаточно простые корреляции D(dм) можно получить только для диффузии простейших газов (одно- и двухатомных с низкой критической температурой) [5].
Попытки решить указанные выше проблемы предпринимались с помощью фрактального анализа на примере горных пород [24; 25]. Для полимеров применение такого подхода имеет достаточно очевидные причины. Во-первых, как отмечалось выше, структура твердых полимеров представляет собой фрактальный объект. Во-вторых, фрактальная размерность отражает оба указанных выше фактора: увеличение этой размерности означает уменьшение и t, и bм [26; 27]. Таким образом, фрактальный анализ представляет возможность теоретического прогнозирования диффузионных характеристик [22].
В работе [5] приведены величины D для ПЭНП и ПЭВП и нескольких газообразных пенетрантов: N2, O2, CO, CO2 и HCl. Для теоретического описания D авторы [22] использовали уравнение, приведенное в работе [24] для анализа диффузии в горных породах:
![]() , (2.5)
, (2.5)
где - константа, которая в дальнейшем принимается равной величине D для полимерного расплава ( = 3,8 ´ 10-7 см2/с), что оказалось универсальным значением, F - пористость, l1 и l2 - верхний и нижний пределы фрактального поведения, соответственно, D¢ - фрактальная размерность, контролирующая процессы газопереноса.
Рассмотрим определение параметров уравнения (2.5) применительно к полимерам. В качестве F для них естественно использовать флуктуационный свободный объем fc, через микрополости которого и происходит диффузия газов [28]. Величина fc может быть оценена из следующего уравнения [6]:
fc = 0,113 jа. (2.6)
В качестве l1 и l2 следует принять диаметры микрополости свободного объема dh и молекулы газа-пенетранта dм, cоответственно. Объем микрополости флуктуационного свободного объема nh можно рассчитать так [29]:
![]() , (2.7)
, (2.7)
где nр - коэффициент Пуассона, k - постоянная Больцмана, Е - модуль упругости. Полагая, что микрополость имеет сферическую форму, из известных величин nh можно рассчитать dh. Величины dм в дальнейшем принимались согласно литературным источникам [5; 30; 31]. Поскольку процесс диффузии газов реализуется на молекулярном уровне [9], то в качестве ds приняты величины спектральной размерности полимерной цепи. Для разветвленного ПЭНП ds = 1,33, для линейного ПЭВП - ds = 1,0 [32]. Необходимые для расчета величин D параметры оцененные указанными методами (Df рассчитана согласно (1.25), df - согласно (1.41), приведены в табл. 2.1.
Таблица 2.1
Параметры фрактальной модели диффузии газов в полиэтиленах [22]
|
Полимер |
fc |
nh, Å3 |
dп, Å |
Df |
ds |
|
ПЭНП |
0,060 |
226 |
7,56 |
3,72 |
1,33 |
|
ПЭВП |
0,024 |
50,5 |
4,58 |
4,13 |
1,0 |
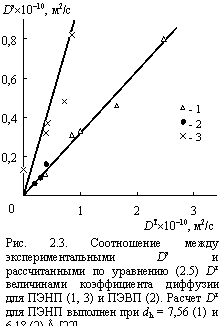
На рис. 2.3 приведено сравнение рассчитанных по уравнению (2.5) Dт и полученных экспериментально [5] Dэ величин коэффициента диффузии для ПЭНП и ПЭВП. Как можно видеть, получено превосходное качественное соответствие, но величины Dт примерно в 3 раза превышают экспериментальные значения Dэ. Основной причиной указанного количественного несоответствия являются разные образцы полиэтиленов, использованные для оценки механических свойств (уравнения (1.14) и (1.15)) и измерений D. Кроме того, фактором, увеличивающим погрешность, является типичная для фрактальных соотношений сильная степенная зависимость D, описываемая уравнением (2.5) [22].
Рассмотрим эти факторы на примере ПЭНП, для которого анализ размеров микрополости свободного объема методом аннигиляции позитронов дал величину наиболее продолжительной компоненты долговечности позитрона t3 = 2,2 нс при Т = 293 К [33]. В работе [34] приведена упрощенная формула, позволяющая оценить величину nh по известным значениям t3:
t3 = 7,80nh + 1,29. (2.8)
Оценка по уравнению (2.8) дает nh = 124Å3 или dh = 6,18 Å. Отметим, что образцы ПЭНП, использованные для измерений D [5] и t3 [33] имели близкие значения плотности r (0,914 и 0,912 г/см3, соответственно) и, следовательно, близкие величины К, что дает основания для их сравнения. Расчет величин DT для ПЭНП с использованием dh = 6,18 Å вместо использованного ранее dh = 7,56 Å (табл. 2.1) дает гораздо лучшее соответствие между Dт и Dэ (рис. 2.3). Еще одним источником завышения величин Dт может быть погрешность оценки nр согласно соотношению (1.15) [35].
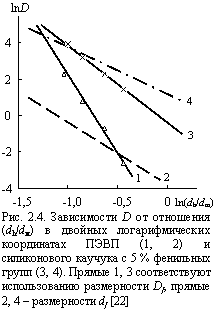
Для оценки величины D согласно уравнению (2.5) авторы [22], использовали размерность Df в качестве D¢, а не фрактальную размерность структуры df. Формальная причина этого заключается в том, что Df контролирует свойства флуктуационного свободного объема [36] и также следующее экспериментальное наблюдение. Как следует из уравнения (2.5), для одного и того же полимера величины , fc, D1 и l1 являются постоянными и поэтому зависимость D от (dh/dм) (или 1/dм) в двойных логарифмических координатах должна быть прямой с наклоном 2(D¢ - ds)/ds. На рис. 2.4 приведена такая зависимость для ПЭВП, где величины D взяты для ряда инертных газов [5]. Как и ожидалось, получена линейная зависимость, указывающая на быстрый спад D по мере увеличения dм. наклон этой зависимости D » 8, что дает величину D¢ = 5 при ds = 1,0. Поскольку максимальная величина df = d = 3 [37], то наклон зависимости D(dh/dм) в двойных логарифмических координатах при использовании этой размерности не может превышать 4 для линейных полимеров и ~ 2,51 - для разветвленных. Таким образом, использование df для ПЭВП не согласуется с экспериментальными результатами. В то же время величины Df и df однозначно связаны соотношением (1.41). Исходя из D » 8 и Df = 5 (рис. 2.4), получим для ПЭВП согласно уравнению (1.41) df » 2,75 и nр » 0,375, что превосходно согласуется с оценками, сделанными другими методами [29].
Тем не менее, как показано в [26; 27] зависимость коэффициента проницаемости Р от (1/dм) может резко изменить свой наклон. Такое изменение наблюдалось и ранее и было объяснено изменением подвижности цепей [23]. В предлагаемой трактовке это означает, что при dм < ![]() (где
(где ![]() - некоторое критическое значение dм [26]) взаимодействие молекул газа-пенетранта со стенками микрополостей свободного объема невелико или отсутствует и тогда процесс газопереноса контролируется размерностью df. При dм >
- некоторое критическое значение dм [26]) взаимодействие молекул газа-пенетранта со стенками микрополостей свободного объема невелико или отсутствует и тогда процесс газопереноса контролируется размерностью df. При dм > ![]() на процесс газопереноса сильно влияет взаимодействие молекул газа со стенками микрополостей, которые представляют собой поверхность макромолекул с высокой молекулярной подвижностью вследствие расстеклованного состояния аморфной фазы полиэтиленов и, следовательно, высоким значением Df. Иначе говоря, при dм <
на процесс газопереноса сильно влияет взаимодействие молекул газа со стенками микрополостей, которые представляют собой поверхность макромолекул с высокой молекулярной подвижностью вследствие расстеклованного состояния аморфной фазы полиэтиленов и, следовательно, высоким значением Df. Иначе говоря, при dм < ![]() газоперенос реализуется через систему микрополостей, сформированных совокупным действием аморфной и кристаллической фаз, и процесс контролируется размерностью df. При dм <
газоперенос реализуется через систему микрополостей, сформированных совокупным действием аморфной и кристаллической фаз, и процесс контролируется размерностью df. При dм < ![]() основным механизмом газопереноса является взаимодействие молекул газа со стенками микрополостей расстеклованной аморфной фазы, поэтому процесс контролируется размерностью Df. По существу, в процессе газопереноса наблюдается два разных режима для dм <
основным механизмом газопереноса является взаимодействие молекул газа со стенками микрополостей расстеклованной аморфной фазы, поэтому процесс контролируется размерностью Df. По существу, в процессе газопереноса наблюдается два разных режима для dм < ![]() и dм >
и dм > ![]() : структурный и молекулярный, соответственно [26; 27]. К вопросу взаимодействия газ - полимер мы неоднократно будем обращаться ниже вследствие важности этого фактора.
: структурный и молекулярный, соответственно [26; 27]. К вопросу взаимодействия газ - полимер мы неоднократно будем обращаться ниже вследствие важности этого фактора.
В уравнении (2.5) размер молекул газа выражен их диаметром dм, хотя для этой же цели используются и другие параметры: молекулярная масса М [4] или мольный объем [38]. В работе [4] использована следующая зависимость D от размера молекул газа-пенетранта:
![]() , (2.9)
, (2.9)
где kм и bм - константы.
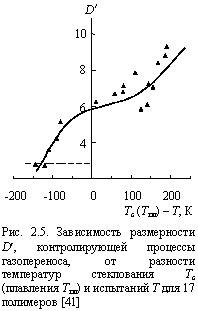
Авторы работы [40] обнаружили V-образную зависимость D от величины Тс, соответствующую по форме зависимости частоты корреляции парамагнитных зондов nс от Тс. На основе этого соответствия был сделан вывод, что увеличение D обусловлено ростом относительного свободного объема про мере повышения Тс. Однако и это объяснение носит качественный характер. Авторы [41] объяснили наблюдаемые зависимости D(Tc) в рамках одной количественной трактовки, а именно, фрактальной модели процессов газопереноса в полимерах [22]. Отметим, что авторы [41] использовали модифицированный критерий выбора размерности в уравнении (2.5): при (dh/dм) > 1,70 в качестве D¢ выбирается df, а при (dh/dм) £ 1,70 - Df.
Нетрудно видеть, что уравнения (2.5) и (2.9) дают одинаковую функциональную форму зависимости D от размера молекул газа-пенетранта: в силу степенной зависимости величина D быстро снижается по мере роста dм или М. Однако, уравнение (2.5) не содержит эмпирических (подгоночных) параметров, что позволяет использовать его для прогнозирования D. На рис. 2.5 приведена зависимость D¢ от разности температур стеклования Тс (или плавления Тпл для аморфно-кристаллических полимеров) и испытаний Т для 17 полимеров. Как и в случае зависимости bм(Тс - Т) [39], наблюдается сигмоидальная зависимость D¢[Tc(Tпл) - Т), хотя и несколько иной формы. Горизонтальные штриховые линии показывают предельные величины D¢ для реальных полимеров: df = 2,95 и Df = 10,1 [42]. Как отмечалось выше, величина t3 характеризует размер микрополости свободного объема [34]. На рис. 2.6 приведена зависимость t3 от разности [Tc (Tпл) - Т] для 13 полимеров. Как и ранее [39; 43], получена V-образная зависимость с минимумом t3 при Тс = T (Tпл = Т). Эта зависимость позволяет прогнозировать величину объема микрополости свободного объема nh согласно уравнению (2.8).

И последнее замечание необходимо сделать относительно выбора величины dh. Как было показано в работе [44], при расчете D по уравнению (2.5) следует учитывать не номинальный диаметр микрополости dh, а его эффективное значение . Это обусловлено тем, что стенки микрополости представляют собой шероховатые на атомном уровне поверхности сегментов макромолекул, колеблющихся вокруг своего положения равновесия [45; 46]. Условие D¢ = df означает, что молекула газа-пенетранта не взаимодействует со стенками микрополости и тогда в качестве принимается [44]:
![]() = dh + Ddh, (2.10)
= dh + Ddh, (2.10)
где Ddh - удвоенная амплитуда колебаний сегментов, окружающих микрополость.
При D¢ = Df указанное взаимодействие реализуется и тогда [44]:
![]() = dh - Ddh. (2.11)
= dh - Ddh. (2.11)

![]() , (2.12)
, (2.12)
где а - нижний линейный масштаб фрактального поведения, rh - радиус микрополости свободного объема, dп - размерность поверхности стенки микрополости.
Величина а принимается равной длине скелетной связи цепи полимера l0, поскольку при а < l0 макромолекула теряет свою индивидуальность [48], а величина dп может быть оценена так [49]:
dп = df - 0,14. (2.13)
Таким образом, теперь получен полный набор параметров уравнения (2.5) (fc, , dм [30], D¢ и ds = 1,0), что позволяет прогнозирование величины D для произвольного линейного полимера. Для сшитых (разветвленных) полимеров следует использовать ds = 1,33. В работе [40] были получены эмпирические аппроксимации, позволяющие описать зависимость D от Тс. В случае диффузии О2 они имеют вид [40]:
lgD = - (3,44 + 0,013 Tc) для Т £ 298К (2.14)
и
lg D = - (9,4 - 0,0058 Tc) для Т ³ 298К. (2.15)
На рис. 2.7 приведено сравнение зависимостей lgD от Тс (или Тпл для диффузии О2, рассчитанных по уравнениям (2.14) и (2.15) (прямые линии) и уравнению (2.5) (точки). Как можно видеть, получено хорошее соответствие обоих наборов данных, причем расчет по уравнению (2.5) по точности примерно соответствует экспериментальному определению D [40].
В табл. 2.2 приведено сравнение экспериментальных и рассчитанных по уравнению (2.5) величин D для пяти полимеров и трех газов (Ar, N2, O2). Как можно видеть, получено удовлетворительное соответствие теории и эксперимента, которое подтверждает корректность предложенной трактовки.
Таким образом, предложенная фрактальная модель адекватно объясняет зависимость коэффициента диффузии от температуры стеклования (плавления) полимеров. Поскольку при расчете величины D в рамках фрактальной модели используются только структурные параметры, определяемые независимыми методами, то появляется возможность прогнозирования величины D с помощью двух калибровочных графиков. Такой расчет дает удовлетворительное соответствие теории и эксперимента [41].
Как известно [4; 5; 21], растворимость газов играет важную роль в процессах переноса для полимеров, в значительной степени определяя их газопроницаемость. Авторы работы [31] предложили следующее корреляционное соотношение для описания коэффициента растворимости газов s в полимерах:
lgs = K1 + K2(e/k)эф, (2.16)
где K1 и K2 - изотермические константы для данного полимера, (e/k)эф - характеристическая силовая постоянная потенциала Леннарда-Джонса, рассчитанная для взаимодействия газ - газ.
Таблица 2.2
Сравнение экспериментальных и рассчитанных по уравнению (2.5) коэффициентов диффузии D [ 41]
|
Полимер |
D, см2/с |
|
|
Расчет по уравнению (2.5) |
эксперимент |
|
|
Полидиметилсилоксан |
109 |
108 |
|
79 |
94 |
|
|
107 |
115 |
|
|
Поливинилтриметилсилан |
7,1 |
4,8 |
|
3,9 |
3,6 |
|
|
9,9 |
7,6 |
|
|
Поликарбонат |
2,9 |
0,2 |
|
1,6 |
1,3 |
|
|
5,3 |
0,2 |
|
|
Поливинилацетат |
0,05 |
0,16 |
|
0,04 |
0,06 |
|
|
0,07 |
0,50 |
|
|
Натуральный каучук |
5,8 |
13,6 |
|
5,3 |
11,7 |
|
|
7,5 |
17,3 |
|
Соотношение (2.16) позволяет достаточно точный расчет величины s [31], но наличие двух эмпирических констант, тем более различающихся для разных температур, делает прогнозирование величины s согласно этому методу затруднительным. Кроме того, в уравнении (2.16) отсутствуют какие-либо структурные характеристики полимера, хотя они могут входить в константы K1 и K2 неявным образом. В работах [45; 46] была предложена новая фрактальная модель микрополости флуктуационного свободного объема, которая отличается от традиционных, вкратце описанная выше. Показано, что микрополость моделируется Df-мерной (а не трехмерной) сферой и размерность ее стенок примерно равна Df, т.е., связана с энергетическими характеристиками структуры [50]. На основе этой трактовки авторы работы [51] предложили фрактальную модель для количественного описания растворимости газов с учетом как структуры полимеров, так и структурных характеристик растворяющихся газов.
Предполагается, что растворимость простых газов, не взаимодействующих с полимером, реализуется осаждением их молекул на стенках микрополостей флуктуационного свободного объема, т.е., рассматривается «истинная» сорбция [23]. В этом случае можно записать [52]:
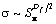 , (2.17)
, (2.17)
где Sм - поперечное сечение молекулы растворяющегося газа.
Поскольку для серии углеводородов С1 - С4, рассмотренных в [51], предполагается наличие взаимодействия между молекулами растворяющегося газа [31], то этот фактор может быть охарактеризован с помощью силовой постоянной потенциала Леннарда-Джонса, рассчитанной именно для таких взаимодействий (см. табл. 2.3). Тогда фрактальную формулу для расчета s можно записать в следующем виде [51]:
![]() , (2.18)
, (2.18)
где s0 - константа, ![]() - эффективная площадь поперечного сечения молекулы растворяющегося газа, учитывающая возможность различных ориентацией этой молекулы в микрополости свободного объема.
- эффективная площадь поперечного сечения молекулы растворяющегося газа, учитывающая возможность различных ориентацией этой молекулы в микрополости свободного объема.
Таблица 2.3
Величины диаметров dм, эффективных для процессов растворимости диаметров и силовых постоянных (e/k) для углеводородов С1 - С4 [51]
|
Углеводород |
dм, Å [31] |
, Å [31] |
, Å, уравнение (2.1.9) |
(e/k), K [31] |
|
CH4 |
3,3 ¸ 4,2 |
3,18 ± 0,14 |
3,30 |
154,7 ± 15,6 |
|
C2H6 |
3,7 ¸ 5,2 |
3,69 ± 0,08 |
4,06 |
250 ± 22 |
|
C3H8 |
4,1 ¸ 5,8 |
4,09 ± 0,08 |
4,76 |
305 ± 28 |
|
C4H10 |
4,6 ¸ 6,9 |
4,40 ± 0,08 |
5,66 |
364 ± 35 |
|
C2H4 |
3,6 ¸ 5,5 |
3,57 ± 0,08 |
3,82 |
225 ± 15 |
|
C3H6 |
4,1 ¸ 5,4 |
3, 68 ± 0,13 |
4,63 |
294 ± 32 |
|
C4H8 - 1 |
4,5 ¸ 5,2 |
4,18 |
5,36 |
356 |
|
C2H2 |
3,5 ¸ 5,7 |
3,38±0,06 |
3,70 |
223 ± 23 |
|
C3H4 (м) |
4,0 ¸ 4,8 |
3,62±0,07 |
4,62 |
321 ± 25 |
|
C4H6 (э) |
4,4 ¸ 5,0 |
3,81 |
5,26 |
360 |
|
С3Н4 (а) |
4,6 ¸ 4,9 |
3,64±0,07 |
5,38 |
335 ± 14 |
|
С4Н6 (б) |
5,0 |
3,85 |
5,45 |
327 |
Примечание: м - метилацетилен, э - этилацетилен, а - аллен, б - бутадиен.
В справочной литературе диаметры dм молекул углеводородов даются с большим разбросом, причем максимальный диаметр ![]() может превышать минимальный
может превышать минимальный ![]() в 1,5 раза (например, для С4Н10, см. табл. 1 работы [31]). По существу, определение эффективного диаметра
в 1,5 раза (например, для С4Н10, см. табл. 1 работы [31]). По существу, определение эффективного диаметра ![]() означает оценку ориентации молекулы газа в микрополости свободного объема. Чтобы сделать расчет s достаточно точным, необходимо уметь прогнозировать величину (и, следовательно, в уравнении (2.18)). В противном случае в силу степенного характера уравнения (2.18) вариация s при изменении dм в 1,5 раза может достигать одного порядка.
означает оценку ориентации молекулы газа в микрополости свободного объема. Чтобы сделать расчет s достаточно точным, необходимо уметь прогнозировать величину (и, следовательно, в уравнении (2.18)). В противном случае в силу степенного характера уравнения (2.18) вариация s при изменении dм в 1,5 раза может достигать одного порядка.

Выполненный обратный расчет, т.е., оценка по известным значениям s показал систематическое увеличение отношения / по мере роста (e/k), что показано на рис. 2.8. Аналитически указанное соотношение может быть записано так [51]:
![]() . (2.19)
. (2.19)
В дальнейшем для расчета s авторы [51] использовали значения ![]() (и, следовательно,
(и, следовательно, ![]() ), оцененные согласно уравнению (2.19).
), оцененные согласно уравнению (2.19).
Отметим, что если размеры ![]() и
и ![]() рассматривать как наибольший и наименьший размеры молекул газа неправильной формы, то увеличение отношения
рассматривать как наибольший и наименьший размеры молекул газа неправильной формы, то увеличение отношения ![]() по мере роста (e/k) означает разворот молекулы растворяющегося газа в микрополости свободного объема наибольшим сечением коллинеарно стенке этой микрополости, что описывает ориентацию молекулы газа в процессе сорбции. Такой разворот или увеличение приводит к росту s согласно формуле (2.18).
по мере роста (e/k) означает разворот молекулы растворяющегося газа в микрополости свободного объема наибольшим сечением коллинеарно стенке этой микрополости, что описывает ориентацию молекулы газа в процессе сорбции. Такой разворот или увеличение приводит к росту s согласно формуле (2.18).
Как показали оценки константы s0 в уравнении (2.18), для полиэтиленов (П) и поливинилтриметилсилана (ПВТМС) она имеет примерно одинаковую величину и равна 4,0 ´ 10-8, а для каучукообразного полиизопрена (ПИ) s0 = 4,8 ´ 10-11, т.е., значительно ниже. Как правило, в уравнениях, подобных формуле (2.18), физический смысл константы определяется при равенстве параметра (в нашем случае s) и константы (в нашем случае s0) при условии, что остальные члены в правой части уравнения равны единице. Для формулы (2.18) это условие означает [51]:
![]() . (2.20)
. (2.20)

Рассмотрим условия реализации критерия (2.20). Минимальное значение dм для гелия равно 1,8 Å [5], что соответствует = 2,54 Å2. Для минимального значения nр = - 1, что соответствует очень пористому твердому телу, Df = 1,33 согласно уравнению (1.25). Тогда для реализации критерия (2.20) необходимо выполнение условия (e/k) = 0,538 К. Эта величина (e/k) примерно на три порядка меньше средней величины (e/k) для углеводородов С1 - С4 [31], т.е., это означает практически полное отсутствие взаимодействий между молекулами растворяющегося газа. Таким образом, величина s0 которая для T < Tc(Tпл) в среднем на 6 ¸ 7 порядков, а для T > Tc(Tпл) даже на 9 ¸ 10 порядков ниже s, определяет минимальную растворимость газа с наименьшим возможным диаметром его молекулы и наименьшей энергией структуры полимера при отсутствии взаимодействий газ - газ. Увеличение dм, Df и (e/k), характеризующих указанные факторы, приводит к резкому росту s [51].

Таким образом, фрактальная модель позволяет достаточно точное количественное описание растворимости газов в полимерах, Предложенное для этой цели уравнение (2.18) по существу показывает увеличение реальной величины s по сравнению с минимально возможной s0. Это увеличение контролируется изменением следующих факторов: размера и ориентации молекул растворяющегося газа, взаимодействия между этими молекулами и структуры полимера. Последний фактор наиболее сильно влияет на величину s в силу степенного характера зависимости этого параметра от Df [51].