
Научная электронная библиотека
Монографии, изданные в издательстве Российской Академии Естествознания

5.2. Структурная трактовка термоокислительной деструкции твердофазных аморфнокристаллических полимеров
В работах [9; 10] было показано, что зависимости относительной ударной вязкости ![]() от продолжительности теплового старения tст для ПБТФ + Z распадаются на три участка (для исходного ПБТФ - на два участка), которые показаны для ПБТФ и двух композиций ПБТФ + Z на рис. 4.21 и 4.22, соответственно. Применение фрактального анализа позволило выяснить физические причины такого поведения функции
от продолжительности теплового старения tст для ПБТФ + Z распадаются на три участка (для исходного ПБТФ - на два участка), которые показаны для ПБТФ и двух композиций ПБТФ + Z на рис. 4.21 и 4.22, соответственно. Применение фрактального анализа позволило выяснить физические причины такого поведения функции ![]() и дать химические механизмы термоокислительной деструкции для каждого из участков (см. раздел 4.4 предыдущей главы). Однако, из графиков рис. 4.21 и 4.22 следует, что для ПБТФ и композиций ПБТФ + Z с разным содержанием Z участок с наиболее быстрым спадом
и дать химические механизмы термоокислительной деструкции для каждого из участков (см. раздел 4.4 предыдущей главы). Однако, из графиков рис. 4.21 и 4.22 следует, что для ПБТФ и композиций ПБТФ + Z с разным содержанием Z участок с наиболее быстрым спадом ![]() по мере роста tст (и, следовательно, нежелательный в практических приложениях ПБТФ) прекращается при разных значениях
по мере роста tст (и, следовательно, нежелательный в практических приложениях ПБТФ) прекращается при разных значениях ![]() , после чего начинается гораздо более медленный спад
, после чего начинается гораздо более медленный спад ![]() , т.е., более предпочтительный с практической точки зрения. Авторы [11; 12] выяснили причины такого поведения зависимости
, т.е., более предпочтительный с практической точки зрения. Авторы [11; 12] выяснили причины такого поведения зависимости ![]() для ПБТФ и композиций ПБТФ + Z, а также получили рекомендации, позволяющие сокращение или подавление участка наиболее быстрого спада указанной функции.
для ПБТФ и композиций ПБТФ + Z, а также получили рекомендации, позволяющие сокращение или подавление участка наиболее быстрого спада указанной функции.
Рассмотрим зависимости ![]() , приведенные на рис. 4.21 и 4.22, несколько подробнее. На первом участке этих зависимостей для композиций ПБТФ + Z, характеризуемом условием
, приведенные на рис. 4.21 и 4.22, несколько подробнее. На первом участке этих зависимостей для композиций ПБТФ + Z, характеризуемом условием ![]() = const = 1,0, реализуется ингибированная термоокислительная деструкция, описываемая в рамках задачи о ловушках [13]. Отсутствие ингибитора термоокислительной деструкции в исходном ПБТФ приводит к отсутствию для него указанного участка. Как отмечалось в предыдущей главе, смесь Fe/FeO является также сильным модификатором структуры, приводящим к экстремальному росту относительной доли кластеров jкл при содержании Z CZ » 0,05 ¸ 0,10 вес. % и соответствующему росту протяженности первого участка (периода индукции)
= const = 1,0, реализуется ингибированная термоокислительная деструкция, описываемая в рамках задачи о ловушках [13]. Отсутствие ингибитора термоокислительной деструкции в исходном ПБТФ приводит к отсутствию для него указанного участка. Как отмечалось в предыдущей главе, смесь Fe/FeO является также сильным модификатором структуры, приводящим к экстремальному росту относительной доли кластеров jкл при содержании Z CZ » 0,05 ¸ 0,10 вес. % и соответствующему росту протяженности первого участка (периода индукции) ![]() . Кроме того, было установлено (см. раздел 4.4 предыдущей главы), что на втором участке зависимости
. Кроме того, было установлено (см. раздел 4.4 предыдущей главы), что на втором участке зависимости ![]() реализуется псевдомономолекуляррная реакция (реакция промежуточных продуктов распада с макромолекулой полимера), а на третьем (втором для ПБТФ) - бимолекулярная реакция (реакция кислорода с макромолекулой) [9].
реализуется псевдомономолекуляррная реакция (реакция промежуточных продуктов распада с макромолекулой полимера), а на третьем (втором для ПБТФ) - бимолекулярная реакция (реакция кислорода с макромолекулой) [9].
Из более внимательного рассмотрения графиков рис. 4.22 можно сделать вывод, что увеличение ![]() сопровождается прекращением псевдомономолекулярной реакции при более высоких значениях
сопровождается прекращением псевдомономолекулярной реакции при более высоких значениях ![]() , т.е., ослаблением эффекта данной реакции. Как отмечалось выше, увеличение
, т.е., ослаблением эффекта данной реакции. Как отмечалось выше, увеличение ![]() обусловлено экстремальным ростом jкл в интервале СZ = 0,05 ¸ 0,10 вес. % и соответствующим ослаблением диффузии кислорода в некристаллические области полимера. Поскольку модифицирующее действие Z сохраняется и после исчерпания его ингибирующего действия, то отмеченное выше соответствие между величиной
обусловлено экстремальным ростом jкл в интервале СZ = 0,05 ¸ 0,10 вес. % и соответствующим ослаблением диффузии кислорода в некристаллические области полимера. Поскольку модифицирующее действие Z сохраняется и после исчерпания его ингибирующего действия, то отмеченное выше соответствие между величиной ![]() и значением
и значением ![]() , при котором происходит переход от псевдомоно- к бимолекулярной реакции, обусловлено теми же эффектами структурной стабилизации. Сокращение участка псевдомономолекулярной реакции по мере роста
, при котором происходит переход от псевдомоно- к бимолекулярной реакции, обусловлено теми же эффектами структурной стабилизации. Сокращение участка псевдомономолекулярной реакции по мере роста ![]() позволяет сделать предположение, что указанная реакция протекает в наименее плотноупакованных областях полимера. В рамках кластерной модели показано [12], что структура аморфно-кристаллического полимера состоит из двух фаз - аморфной и кристаллической, а первая из указанных фаз состоит из кластеров и рыхлоупакованной матрицы. Относительную долю рыхлоупакованной матрицы jр.м. можно определить из соотношения (2.33. В свою очередь, величина jр.м. в расчете на весь полимер (приведенная величина
позволяет сделать предположение, что указанная реакция протекает в наименее плотноупакованных областях полимера. В рамках кластерной модели показано [12], что структура аморфно-кристаллического полимера состоит из двух фаз - аморфной и кристаллической, а первая из указанных фаз состоит из кластеров и рыхлоупакованной матрицы. Относительную долю рыхлоупакованной матрицы jр.м. можно определить из соотношения (2.33. В свою очередь, величина jр.м. в расчете на весь полимер (приведенная величина ![]() ) определяется так [14]:
) определяется так [14]:
![]() . (5.1)
. (5.1)
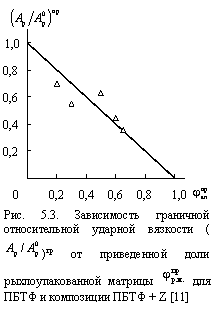
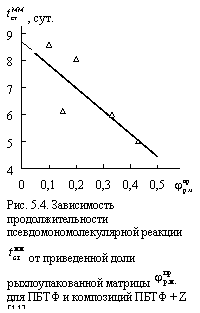
Из сказанного выше можно сделать еще одно предположение, а именно, что увеличение ![]() должно снижать продолжительность действия псевдомономолекулярной реакции. Действительно, приведенная на рис. 5.4 зависимость продолжительности этой реакции
должно снижать продолжительность действия псевдомономолекулярной реакции. Действительно, приведенная на рис. 5.4 зависимость продолжительности этой реакции ![]() от
от ![]() подтверждает это предположение. При
подтверждает это предположение. При ![]() = 0 » 8,7 сут., т.е., для исходного ПБТФ это время является периодом индукции, а затем происходит переход к бимолекулярной реакции. Экстраполяция графика
= 0 » 8,7 сут., т.е., для исходного ПБТФ это время является периодом индукции, а затем происходит переход к бимолекулярной реакции. Экстраполяция графика ![]() (
( ![]() ) к
) к ![]() = 1,0 дает, как и следовало ожидать
= 1,0 дает, как и следовало ожидать![]() = 0. Это означает, что в указанных условиях псевдомономолекулярная реакция протекает очень быстро. Подтверждение этого вывода можно получить из результатов теплового старения ПБТФ при температурах выше его температуры плавления, т.е., теплового старения его расплава, где К = 0 и jкл = 0 и, следовательно, из уравнения (2.33) jр.м. =
= 0. Это означает, что в указанных условиях псевдомономолекулярная реакция протекает очень быстро. Подтверждение этого вывода можно получить из результатов теплового старения ПБТФ при температурах выше его температуры плавления, т.е., теплового старения его расплава, где К = 0 и jкл = 0 и, следовательно, из уравнения (2.33) jр.м. =![]() = 1,0. В этом случае процесс термоокислительной деструкции полностью завершается (полимер практически теряет свою вязкость) за период менее одного часа, что во временном масштабе рис. 5.4 практически означает
= 1,0. В этом случае процесс термоокислительной деструкции полностью завершается (полимер практически теряет свою вязкость) за период менее одного часа, что во временном масштабе рис. 5.4 практически означает ![]() » 0.
» 0.
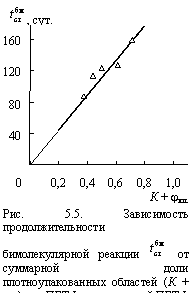
Логическим следствием рассмотренного выше структурного механизма псевдомономолекулярной реакции является предположение, что бимолекулярная реакция представляет собой термоокислительную деструкцию плотноупакованных областей полимера (кристаллитов и кластеров) и именно это обстоятельство определяет резкое снижение скорости ее протекания по сравнению со скоростью псевдомономолекулярной реакции. Чтобы подтвердить это положение, авторы [11] определили продолжительность бимолекулярной реакции ![]() экстраполяцией зависимости
экстраполяцией зависимости ![]() к Ар = 0. Зависимость полученной таким образом величины
к Ар = 0. Зависимость полученной таким образом величины ![]() от суммарной относительной доли плотноупакованных областей (кристаллитов и кластеров) (К + jкл) приведена на рис. 5.5. Как следует из данных этого рисунка, наблюдается линейный рост
от суммарной относительной доли плотноупакованных областей (кристаллитов и кластеров) (К + jкл) приведена на рис. 5.5. Как следует из данных этого рисунка, наблюдается линейный рост ![]() по мере увеличения (К + jкл) и при (К + jкл) = 0 величина
по мере увеличения (К + jкл) и при (К + jкл) = 0 величина ![]() также равна нулю. Это означает, что в отсутствие плотноупакованных областей бимолекулярная реакция не имеет места. Этот вывод подтверждается результатами теплового старения расплава ПБТФ, где термоокислительная деструкция полностью реализуется в рамках псевдомономолекулярной реакции.
также равна нулю. Это означает, что в отсутствие плотноупакованных областей бимолекулярная реакция не имеет места. Этот вывод подтверждается результатами теплового старения расплава ПБТФ, где термоокислительная деструкция полностью реализуется в рамках псевдомономолекулярной реакции.
В главе 4 было продемонстрировано влияние пространственного (структурного) и временного (энергетического) типов беспорядка на процесс термоокислительной деструкции в случае композиций ПЭВП + Z. Ниже мы рассмотрим структурные аспекты этих эффектов.
На рис. 5.6 и 5.7 показаны зависимости концентрации карбонильных групп [> C = 0] Cкарб от продолжительности теплового старения tст при температурах старения Тст = 353 и 463 К, соответственно, для ПЭВП и двух композиций ПЭВП + Z. Как можно видеть, качественно зависимости Скарб(tст) для обеих использованных Тст симбатны и различие по существу заключается в шкале tст или скорости термоокислительной деструкции (сравните с рис. 4.15 и 4.16).

![]() = Cкарб. (5.2)
= Cкарб. (5.2)
Далее можно записать форму зависимости ![]() от времени tст [21]:
от времени tст [21]:
![]() , (5.3)
, (5.3)
по аналогии с соотношением (4.9).

![]() , (5.4)
, (5.4)
где с - некоторая константа.
Рассмотрим структурные аспекты увеличения gв или усиления временного (энергетического) беспорядка. Алгебраическое затухание F(t) в общем случае можно описать так [20]:
![]() , gв > 0, t > t, (5.5)
, gв > 0, t > t, (5.5)
где t - продолжительность реакции, t - время релаксации.
Прологарифмировав соотношение (5.5), получим, что обратная величина gв пропорциональная lnt. В свою очередь, уравнение Дулитла дает взаимосвязь lnt и свободного объема полимера fc [23]:
![]() , (5.6)
, (5.6)
где В - константа.
Величина fc для Тст = 353 К может быть оценена из уравнения [23]:
![]() , (5.7)
, (5.7)
где nр - коэффициент Пуассона.
Для расплава ПЭВП (Тст = 463 К) принята величина fc = 0,113 [24], а для композиций ПЭВП + Z величина fc для расплава рассчитана из уравнения [25]:
![]() , (5.8)
, (5.8)
где h - вязкость расплава, принимаемая равной обратной величине показателя текучести расплава (ПТР) [26], а константа А определена из известных данных fc = 0,113 и ПТР для ПЭВП [17].
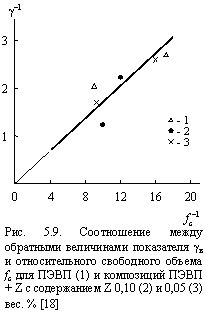
Таким образом, из сочетания уравнений (5.5) ¸ (5.8) следует линейная корреляция между gв и fc. Это положение подтверждается данными рис. 5.9, которые демонстрируют наличие указанной корреляции. Этот факт можно трактовать не только с позиций теории свободного объема, но и с позиций фрактального анализа. Повышение gв по мере роста Тст означает усиление энергетического беспорядка структуры [27]. Как известно [28], степень энергетического возбуждения структуры полимера, а точнее - ее рыхлоупакованных областей, можно охарактеризовать с помощью размерности областей локализации избыточной энергии Df, величина которой определяется согласно уравнению (1.25).
Сочетание уравнений (1.25) и (5.7) позволяет получить следующее соотношение между Df и fc [17]:
![]() . (5.9)
. (5.9)
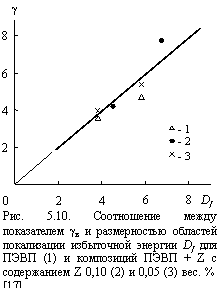
Таким образом, исходя из приведенного на рис. 5.9 соотношения между gв и fc, а также уравнения (5.9), следует ожидать взаимосвязи между gв и Df. Действительно, данные рис. 5.10 подтверждают это предположение, аналитически выражаемое так [17]:
gв » Df. (5.10)
Тогда применительно к термоокислительной деструкции полиэтиленов уравнение (5.3) можно записать следующим образом [18]:
![]() , (5.11)
, (5.11)
где C6 - константа, - ![]() продолжительность периода индукции.
продолжительность периода индукции.
Следовательно, изложенные выше результаты показали, что на кинетику термоокислительной деструкции в полиэтиленах оказывает влияние как пространственный (структурный), так и временной (энергетический) беспорядок. При этом выполняется теорема о субординации, т.е., наблюдается мультипликативное поведение ds и gв (или Df), которые характеризуют эти типы беспорядка. Поскольку тепловое старение обычно выполняется при относительно высоких температурах, то это обстоятельство приводит к повышению роли временного (энергетического) беспорядка.
До сих пор мы рассматривали описание уровня временного (энергетического) беспорядка с помощью показателя gв согласно уравнению (4.16) [20]. Однако, этот эффект может быть рассмотрен и в рамках модели [27; 29], где он учитывается с помощью эффективной спектральной размерности или показателя неоднородности реакционной среды h (см. уравнения (1.68) ¸ (1.70)). По своему физическому содержанию указанные модели идентичны, что позволяет ожидать сходных зависимостей их основных параметров от структурных характеристик. Авторы [30] исследовали соотношение параметра b¢ в уравнении (1.70), характеризующего временной (энергетический) беспорядок [27], и структуры для ряда полимеров.

Поскольку процесс термоокислительной деструкции тесно связан с процессами диффузии кислорода на фрактальной структуре полимеров, а последние контролируются характеристиками свободного объема (см. главу 2), то можно ожидать взаимосвязи fc и b¢ в уравнении (1.70). На рис. 5.11 приведена такая корреляция для пяти полимеров, из которой следует, что зависимость b¢(fc) линейна и проходит через начало координат. Максимальная величина = 2, как следует из уравнения (1.69), и тогда максимальная величина b¢ при условии ds = 1,33 (для разветвленного полимера [22]) равна ~ 1,50.Максимальное значение fc при температурах выше температуры стеклования (плавления) варьируется в пределах 0,109 ¸ 0,159 для разных полимеров [31]. Соответствующие указанным предельным значениям b¢ и fc линейные граничные зависимости b¢(fc) показаны на рис. 5.11 штриховыми линиями. Как можно видеть, экспериментальная зависимость b¢(fc) хорошо согласуется с указанными предельными случаями. Таким образом, данные рис. 5.11 позволяют получить следующую эмпирическую корреляцию [30]:
b¢» 10,3fc. (5.12)
и соотношение (4.16) преобразуется к виду (вновь при условии ds = const = 1,0) [30]:
![]() . (5.13)
. (5.13)
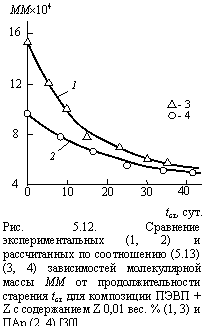
Сравнение экспериментальных и рассчитанных по уравнению (5.13) изменений ММ как функции tст для двух полимеров (ПАр и ПЭВП + Z с содержанием Z 0,01 вес. %) приведено на рис. 5.12, откуда следует хорошее соответствие теории и эксперимента.
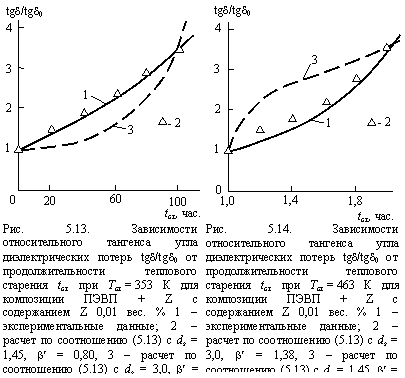
Предложенная трактовка позволяет объяснить большое (примерно на два порядка) различие интенсивностей старения полиэтилена при температурах ниже и выше его температуры плавления Тпл (см. рис. 4.15 и 4.16, а также 5.6 и 5.7). Количественное соответствие может быть получено в рамках единой модели (соотношение (4.16) с заменой gв ds на ) соответствующим выбором величины . Так, при температуре старения Тст < Тпл, равной 353 К, ds = 1,45 и b¢ = 0,80 для композиции ПЭВП = Z, содержащей 0,01 вес. % Z. При Тст > Тпл, равной 463 К, расплав представляет собой каучукоподобное состояние, чья структура является евклидовым (или близким к евклидовому) объектом и для ее ds = 3 и b¢ = 1,38, как отмечалось выше. На рис. 5.13 и 5.14 приведено сравнение экспериментальных и рассчитанных по соотношению (5.13) зависимостей относительного тангенса угла диэлектрических потерь tgd/tgd0 от продолжительности теплового старения tст для композиции ПЭВП + Z с содержанием Z 0,01 вес. % при Тст = 353 и 463 К, соответственно. Как можно видеть, несмотря на большое различие интервалов tст, в обоих случаях получено хорошее соответствие теории и эксперимента. Таким образом, резкое повышение интенсивности термоокислительной деструкции Тст > Тпл обусловлено потерей структурой полимера фрактальных свойств [30]. Отметим, что константа сu в соотношении (5.13) для графиков рис. 5.13 и 5.14 получена методом совмещения (как и для графиков рис. 5.12). При этом для условий Тст < Тпл (рис. 513) и Тст > Тпл (рис. 5.14) указанная константа имеет разные абсолютные величины. На первый взгляд может показаться, что полученное на рис. 5.13 и 5.14 хорошее соответствие теории и эксперимента может быть достигнуто только соответствующим выбором константы cu. Для проверки этого предположения авторы [30] построили теоретические зависимости tgd/tgd0(tст) с показателями соотношения (5.13) наоборот (т.е., для Тст < Тпл ds = 3, b¢ = 1,38 и для Тст > Тпл ds = 1,45, b¢ = 0,80) и соответствующими другими константами cu. Эти зависимости показаны на рис. 5.13 и 5.14 штриховыми линиями. Как можно видеть, в таком варианте соответствия теории и эксперимента получить не удалось. Особенно это заметно для графиков рис. 5.14, где теоретическая и экспериментальные кривые tgd/tgd0(tст) имеют выпуклости разного знака («вверх» и «вниз», соответственно). Следовательно, соответствие теории и эксперимента на графиках рис. 5.13 и 5.14 обусловлено корректным выбором параметром ds и b¢ [30].
Как указано выше, в рамках фрактального анализа степень энергетического беспорядка может быть описана с помощью размерности Df. Отметим, что именно эта размерность контролирует распределение размеров микрополостей флуктуационного свободного объема [32]. Уравнение (5.9) демонстрирует взаимосвязь параметров Df и fc, а сочетание уравнений (5.9) и (5.12) позволяет получить следующее соотношение между b¢ и Df [30]:
b¢ = 0,0876 (3Df - 4). (5.14)
Уравнение (5.14) показывает увеличение b¢ по мере роста степени энергетического беспорядка, характеризуемой размерностью Df.
Сочетание уравнений (5.10) и (5.14) также позволяет получить простое аналитическое соотношение параметров gв и b¢, характеризующих уровень временного (энергетического) беспорядка системы:
gв » 5,14b¢, (5.15)

Связь параметра b¢ с уровнем временного беспорядка можно продемонстрировать следующим образом. При определенном типе распределения плотности энергетических состояний показатель b¢ определяется так [20]:
![]() , (5.16)
, (5.16)
где Т0 - характеристическая температура, описывающая спад плотности энергетических состояний.
Таблица 5.1
Сравнение температур переходов и характеристических температур для ПК и ПАр [30]
|
Полимер |
Т0, К |
Тс¢, К |
Ткр, К |
Tll, К |
|
ПК |
369 |
373 |
509 |
508 |
|
ПАр |
411 |
423 |
567 |
568 |
Из данных рис. 5.11 следует, что для ПК и ПАр коэффициент в уравнении (5.12) несколько меньше средней величины 10,3 и равен ~ 9. Тогда из уравнения (5.12) для b¢ = 1 получим fc » 0,111. На рис. 5.15 показаны зависимости fc(T) для ПК и ПАр, из которых можно оценить температуру Т0, соответствующую условию fc = 0,111 (или b¢ = 1). Эти величины Т0 приведены в табл. 5.1. Как известно [12], теория двухстадийного стеклования полимеров предполагает, что при некоторой температуре ( < Tc) происходит распад нестабильных кластеров, удерживающих рыхлоупакованную матрицу в стеклообразном состоянии, что определяет ее расстекловывание. Обычно принимается следующее приближенное соотношение между Тс¢ и Тс [33]:
Тс¢ = Тс - 50 К. (5.17)

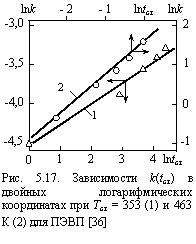
На рис. 5.16 показано сравнение величин b¢ (расчет по уравнению (5.12)) и Т/Т0 для ПК и ПАр. Как можно видеть, получено приблизительное равенство этих параметров, что подтверждает сделанное выше предположение: величина b¢ определяется уровнем временного (энергетического) беспорядка системы [30].
Авторы [36] использовали модель [27; 29] для количественной оценки степени влияния пространственного и временного беспорядка на скорость термоокислительной деструкции ПЭВП. Для экспериментального определения показателя неоднородности h согласно уравнению (1.68) была определена скорость изменения k относительного тангенса угла диэлектрических потерь tgd/tgd0 (см. рис. 4.15 и 4.16) как функция k(tст). На рис. 5.17 приведены зависимости k(tст) в двойных логарифмических координатах, которые позволяют определить величину h из наклона линейных графиков согласно уравнению (1.69). Эти значения приведены в табл. 5.2, откуда следует их существенное различие: Тст = 353 и 463 К. Если для Тст = 353 К величина h относительно невелика и близка к соответствующей величине для евклидовых (гомогенных) сред, то при Тст = 463 К этот показатель близок к величине для полностью неупорядоченных сред. Далее можно рассчитать величину ![]() согласно уравнению (1.69) для обеих Тст, которые также приведены в табл. 5.2. Переход от аморфно-кристаллического состояния (Тст = 353 К) к расплаву (Тст = 463 К) приводит к резкому снижению
согласно уравнению (1.69) для обеих Тст, которые также приведены в табл. 5.2. Переход от аморфно-кристаллического состояния (Тст = 353 К) к расплаву (Тст = 463 К) приводит к резкому снижению ![]() , обусловленному ростом степени гетерогенности последнего. Расчет спектральной размерности ds, характеризующей связность объекта, для твердофазного полимера можно выполнить согласно уравнению (4.29) и тогда ds = 1,46, т.е., практически точно совпадает с величиной . Это указывает, что при Тст = 353 К, т.е., при Тст < Тпл, процесс термоокислительной деструкции контролируется только пространственным (структурным) беспорядком. Для расплава ds можно принять равной соответствующей размерности для линейной макромолекулы, т.е., ds = 1,0 [22], а структурную величину
, обусловленному ростом степени гетерогенности последнего. Расчет спектральной размерности ds, характеризующей связность объекта, для твердофазного полимера можно выполнить согласно уравнению (4.29) и тогда ds = 1,46, т.е., практически точно совпадает с величиной . Это указывает, что при Тст = 353 К, т.е., при Тст < Тпл, процесс термоокислительной деструкции контролируется только пространственным (структурным) беспорядком. Для расплава ds можно принять равной соответствующей размерности для линейной макромолекулы, т.е., ds = 1,0 [22], а структурную величину ![]() (
( ![]() )можно оценить из уравнения (1.72). Тогда
)можно оценить из уравнения (1.72). Тогда ![]() = 0,96 и близка к принятой выше ds (табл. 5.2). Определить величину h, соответствующую пространственному беспорядку в расплаве, можно из уравнения (1.69) при условии
= 0,96 и близка к принятой выше ds (табл. 5.2). Определить величину h, соответствующую пространственному беспорядку в расплаве, можно из уравнения (1.69) при условии ![]() =
= ![]() (h = 0,520).
(h = 0,520).
Таблица 5.2
Сравнение характеристик термооокислительной деструкции ПЭВП ниже (353 К) и выше (463 К)
температуры плавления [36]
|
Тст, К |
h |
|
ds |
|
nпр |
nвр |
n |
|
353 |
0,263 |
1,47 |
1,46 |
1,46 |
2,36 |
1,0 |
2,36 |
|
463 |
0,905 |
0,19 |
1,0 |
0,96 |
3,08 |
3,47 |
11,49 |
Как показано в работе [37], порядок реакции n связан с показателем h следующим уравнением:
![]() . (5.18)
. (5.18)
Расчет порядка реакции nпр, обусловленного пространственным беспорядком, согласно уравнению (5.18) при h = 0,293 Тст = 353 К и h = 0,520 согласно уравнению (1.69) и условию ![]() =
= ![]() для Тст = 463 К показал, что эти значения nпр равны 2,36 и 3,08, соответственно. Иначе говоря, повышение степени пространственного беспорядка, характеризуемое увеличением h от 0,263 до 0,520, приводит к росту nпр от 2,36 при Тст = 353 К до 3,08 при Тст = 463 К. Поскольку при Тст = 353 К
для Тст = 463 К показал, что эти значения nпр равны 2,36 и 3,08, соответственно. Иначе говоря, повышение степени пространственного беспорядка, характеризуемое увеличением h от 0,263 до 0,520, приводит к росту nпр от 2,36 при Тст = 353 К до 3,08 при Тст = 463 К. Поскольку при Тст = 353 К ![]() = ds, т.е., временной беспорядок отсутствует, то общий порядок реакции n равен пространственному: n = nпр. Для второй из указанных температур теплового старения ds = 1 и
= ds, т.е., временной беспорядок отсутствует, то общий порядок реакции n равен пространственному: n = nпр. Для второй из указанных температур теплового старения ds = 1 и ![]() = 0,19, т.е., ds ≠
= 0,19, т.е., ds ≠ ![]() , и величина n, рассчитанная согласно уравнению (5.18), равна 11,49. Отметим, что отношение величин n для Тст = 353 и 463 К равно 4,88, т.е., примерно 5, что соответствует приведенным выше данным [38] (см. главу 4). Очевидно, что при Тст > Тпл порядок реакции n контролируется как пространственным (nпр), так и временным (nвр) беспорядком, что соответствует известным данным [27] и определяется теоремой о субординации [20]:
, и величина n, рассчитанная согласно уравнению (5.18), равна 11,49. Отметим, что отношение величин n для Тст = 353 и 463 К равно 4,88, т.е., примерно 5, что соответствует приведенным выше данным [38] (см. главу 4). Очевидно, что при Тст > Тпл порядок реакции n контролируется как пространственным (nпр), так и временным (nвр) беспорядком, что соответствует известным данным [27] и определяется теоремой о субординации [20]:
N = nпрnвр. (5.19)
Из уравнения (5.19) следует nвр = 3,73, т.е., временной беспорядок оказывает даже большее влияние на величину n при Тст = 463 К, чем пространственный [36].