
Научная электронная библиотека
Монографии, изданные в издательстве Российской Академии Естествознания

ПАРАТГОРМОН-РОДСТВЕННЫЙ ПРОТЕИН. 2-е издание переработанное и дополненное
Курзанов А. Н., Ледванов М. Ю., Быков И. М., Медведев В. Л., Стрыгина Е. А., Бизенкова М. Н., Заболотских Н. В., Ковалев Д. В., Стукова Н. Ю.,
13.1.3.6. Паратгормон-родственный протеин при воспалении и фиброзе почек
Воспаление включает в себя сложные молекулярные и клеточные механизмы, которые активируются против повреждающих факторов и обычно служат положительной биологической цели, но оно может иметь пагубные последствия при хронических патологических состояниях, таких как прогрессирующие заболевания почек (Chevalier R.L., 2006; Rees A.J., 2006; Remuzzi G., Bertani T., 1998). Воспаление играет ключевую роль в прогрессирующем рубцевании и фиброгенезе при различных заболеваниях почек (Remuzzi G., Bertani T.N., 1998; Chevalier R.L., 2006; 2: 157–168; Rees A.J., 2006; Strutz F., Neilson E.G., 2003). Тубулоинтерстициальное воспаление является ключевым событием при различных нефропатиях. Известно, что тубулоинтерстициальное воспаление способствует интерстициальному фиброзу и прогрессированию повреждения почек (Strutz F., Neilson E.G., 1994; Strutz F., Neilson E.G., 2003). После повреждения почек тубулоэпителиальные клетки начинают сверхэкспрессировать провоспалительные цитокины и хемокины, которые способствуют миграции моноцитов/макрофагов и Т-лимфоцитов в почечный интерстиций (Muller G.A. et al., 1992; Rees A.J., 2006). Как инфильтрирующие лейкоциты, так и поврежденные тубулоэпителиальные клетки активируют и индуцируют пролиферацию резидентных фибробластов в тубулоинтерстициальном компартменте. Тяжелое и длительное повреждение определяет устойчивую активацию провоспалительных путей, связанных с избыточной экспрессией профиброгенных цитокинов тубулоинтерстициальными клетками, что приводит к фиброгенезу и потере почечной функции (Strutz F., Neilson E.G., 2003).
Установлено что ПТГрП может действовать как провоспалительный фактор при различных патофизиологических состояниях (Martin-Ventura J.L. et al., 2003). Показано, что ПТГрП может выступать в качестве важного индуктора провоспалительных цитокинов, а именно фактора некроза опухоли и интерлейкина-6, при полиорганном воспалении (Funk J.L., 2001). Продемонстрировано, что ПТГрП активирует ядерный фактор NF-kB и экспрессию NF-kB-зависимых цитокинов и хемокинов и в том числе IL-6 и хемоаттрактантного белка-1 моноцитов (MCP-1) в различных типах клеток (Guillen C. et al., 2002; Martin-Ventura J.L. et al., 2003). Показано, что NF- κB -связанные факторы, по-видимому, играют важную роль в воспалении, связанном с повреждением почек (Gomez-Garre D. et al., 2001; Lee F.T. et al., 2004; Guijarro C., Egido J. 2001; Morrissey J.J., Klahr S. 1997) (рис. 17).
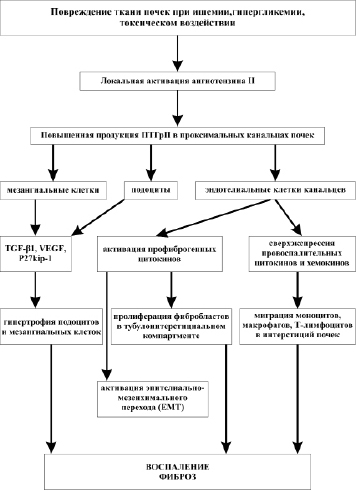
Рис. 17. Эффекты паратгормон-родственного протеина в поврежденной почке при острой почечной недостаточности, диабетической и обструктивной нефропатии
Повреждение мезангиальных клеток (MC) является характерной чертой гломерулонефрита. Активированные MC секретируют медиаторы воспаления, которые вызывают апоптоз клеток. ПТГрП является локально активным полигормоном, который повышает выживаемость клеток и активируется провоспалительными факторами во многих типах клеток. Резудьтаты исследования регуляции экспрессии ПТГрП провоспалительными цитокинами и оценка роли, самогоПТГрП в качестве провоспалительного фактора или фактора выживания мышиных мезангиальных клеток в культуре показали, что IL-1 β и TNF- α быстро и временно повышали экспрессию ПТГрП в MC (Hochane M. et al., 2018). Эффекты IL-1 β были как транскрипционными, так и посттранскрипционными со стабилизацией мРНК ПТГрП человеческим антигеном R. Массивы профиля протеома показали, что сам ПТГрП усиливал цитокины в клеточных лизатах, главным образом, IL-17, IL-16, IL-1 α и IL-6. ПТГрП также стимулировал устойчивую экспрессию хемокинов, в основном регулируемых активацией экспрессируемых и секретируемых нормальных Т-клеток (RANTES)/CC-хемокина 5 (CCL5) и макрофагального воспалительного белка-2 (MIP-2)/CXC-мотива хемокинов 2 (CXCL2)/тимусом и активацией регулируемый хемокин (TARC-thymus and activation regulated chemokine)/CCL17, и индуцируемый интерфероном α-хемоаттрактант Т-клеток (I-TAC)/CXCL11. Более того, ПТГрП заметно усиливал экспрессию циклооксигеназы-2 (ЦОГ-2) и вызывал его автоиндукцию посредством активации пути NF-B. ПТГрП индуцировал выживание мезангиальных клеток через продукты COX-2, а избыточная экспрессия ПТГрП в MC уменьшала апоптотические эффекты IL-1 β и TNF- α. В целом, эти результаты подтверждают, что ПТГрП действует как стимулятор воспалительных процессов в клубочках и может быть компонентом отрицательной обратной связи, сохраняющей выживаемость MC (Hochane M., 2018). Таким образом ПТГрП может способствовать воспалению, пролиферации и выживанию мезангиальных клеток (Hochane M. et al., 2013).
В той же лаборатории исследовали экспрессию и роль ПТГрП в воспалении и заживлении клубочков в экспериментальной модели гломерулонефрита, вызванной внутривенной инъекцией мышам яда змеи Хабу (Hochane M. et al., 2018). Временной анализ показал выраженное повреждение почек в первые дни после введения яда и начало выздоровления в течение 7 дней. Гломерулярная экспрессия ПТГрП (транскрипта и белка) наблюдалась на ранней стадии после введения яда (с 1-го по 3-й день) наряду с воспалительной реакцией. Инактивация секретированного ПТГрП с помощью ПТГрП-нейтрализующего антитела снижала экспрессию маркеров локального воспаления (хемотаксического белка макрофага-1; циклооксигеназы 2; IL-6 и инфильтрации макрофагов) и отменила экспрессию самого ПТГрП. Кроме того, пролиферация клубочковых клеток была заторможена, как и процесс заживления. Эти результаты показали, что ПТГрП обладает антиномическим действием при гломерулонефрите, участвуя как в провоспалительных состояниях, так и в процессе заживления. Эти данные раскрывают существенную роль ПТГрП в раннем восстановлении клубочков в экспериментальной модели гломерулонефрита.
В экспериментах, проведенных (Ramila D. et al., 2008) исследовали функциональные последствия хронической активации ПТГрП в почках мыши после односторонней обструкции мочеточника, характеризующейся ранним воспалительным ответом почек (Chevalier R.L. 2006; Esteban V., Lorenzo O. et al., 2004). Результаты экспериментов с моделированием почечного воспаления на мышах с односторонней обструкцией мочеточника–показали критическую роль ПТГрП в этом состоянии (Ramila D. et al., 2008). Было обнаружено, что ПТГрП активируется даже у мышей с повышенной экспрессией ПТГрП. В отличие от предыдущих наблюдений при ишемическом или нефротоксическом повреждении почек (Ramila D. et al., 2008), PTH1R не подавлялся после обструкции мочеточника у мышей. Кроме того установлено, что после обструкции мочеточника ПТГрП активирует некоторые провоспалительные факторы в тубулоэпителиальных клетках и способствует миграции моноцитов/макрофагов. Увеличение нескольких провоспалительных факторов, а именно: белка-хемоаттрактанта моноцитов (МСР)-1, рецептора МСР-1 CCR2 (рецептор хемокинов (мотив С-С) 2), хемокина, выделяемого T-клетками при активации (Regulated on Activation, Normal T-cell Expressed and Secreted), хемоаттрактанта для моноцитов (RANTES), интерлейкина 6 (IL-6) и молекулы клеточной адгезии, присутствующей на мембранах лейкоцитов (ICAM-1) было более значительным, связанным с более выраженным повреждением канальцев, у мышей с повышенной экспрессией ПТГрП, чем у контрольных животных. Индукция экспрессии MCP-1, RANTES, IL-6 и CCR2 с помощью ПТГрП зависела, по-видимому, от активации как сигнального пути ERK (extracellular signal-regulated kinase), так и NF- κB в тубулоэпителиальных клетках. Это убедительно свидетельствует о том, что эти внутриклеточные пути участвуют в механизмах, посредством которых ПТГрП может способствовать воспалению почек. Более того, ERK-опосредованная активация NF-kB, по-видимому, является важным механизмом, посредством которого ПТГрП вызывает воспаление почек. Подавление эффектов повышенной продукции ПТГрП на активацию провоспалительных факторов с помощью антагониста ангиотензин 1 рецептора лозартана, который как ранее было показано, устраняет активацию ПТГрП, вызванную нефротоксическим острым повреждением почек у крыс (Ortega A. et al., 2005) было столь же эффективным, как и влияние ПТГрП (7–34), являющегося антагонистом PTH1R, в ингибировании воспаления в почке мышей с обструкцией мочеточника. Эти данные позволили авторам анализируемого исследования предположить, что ПТГрП может рассматриваться как новый маркер воспаления и потенциальная терапевтическая мишень в поврежденной почке (Ramila D. et al., 2008). Наконец, поскольку устойчивое почечное воспаление тесно связано с фиброгенезом, эти данные указывают на то, что ПТГрП является вероятным провоспалительным и профиброгенным фактором в поврежденной почке.
Прогрессирование хронического заболевания почек характеризуется непрерывным накоплением и отложением внеклеточного матрикса, что приводит к распространенному фиброзу тканей. Считается, что почечный интерстициальный фиброз является общей характеристикой хронических заболеваний почек, приводящих к терминальной стадии почечной недостаточности (Liu Y., 2004). Интерстициальные фибробласты являются основным типом клеток, ответственных за фиброгенез, процесс, в результате которого эти клетки пролиферируют и становятся активированными миофибробластами (Ardura J.A. et al., 2008). Известно, что фиброз почки индуцируется как тубулоэпителиальными, так и инфильтрирующими клетками, а также секрецией матриксных соединений, как активированными фибробластами, так и тубулярными клетками. Повышенный синтез и отложение матрикса, а также потеря целостности тубулярной структуры являются первостепенными событиями на более поздних стадиях фиброгенеза (Fan J.M. et al., 1999).
Тубулоинтерстициальный фиброз является первостепенным событием в прогрессировании хронической почечной патологии. Фиброгенный процесс характеризуется атрофией канальцев и накоплением белков внеклеточного матрикса, включая фибронектин, коллагены
типов I, III, IV и V, а также ламинин (Chevalier R.L., 2006; Nangaku M., 2004). Экспериментальные данные свидетельствуют о том, что приток макрофагов в почечный интерстиций является ключевым шагом в развитии хронического воспаления и последующего интерстициального фиброза в поврежденной почке (Vielhauer V. et al., 2001). Фиброгенез также сопровождается увеличением пролиферации почечных интерстициальных клеток и их трансформацией в миофибробласты, основные эффекторные клетки в этой ситуации (Lewis M.P., Norman J.T., 1997; Sato M. et al., 2003). В поврежденной почке миофибробласты могут образовываться из резидентных фибробластов и из циркулирующих клеток, происходящих из костного мозга, а также посредством эпителиально-мезенхимального перехода (EMT), процесса, в результате которого тубулоэпителиальные клетки становятся продуцирующими матрицу миофибробластами (Kalluri R., Neilson E.G., 2003; 27Liu Y., 2004).
Посредством трансдифференцировки α-SMA-позитивных миофибробласто в эпителиальные клетки почечных канальцев продуцируют ряд воспалительных и фиброгенных цитокинов, таких как трансформирующий фактор роста b1 (TGF-b1) и фактор роста соединительной ткани (CTGF), которые участвуют в дальнейшем развитии почечного интерстициального фиброза (Abbate M. et al., 2002). Трансформирующий фактор роста-b1 является профиброзным регулятором, который может стимулировать эпителиальные клетки канальцев к прохождению эпителиальной мезенхимальной трансдифференцировки, тогда как CTGF был признан одним из важных факторов, которые опосредуют фиброзную активность TGF-b1 (Border W.A., Noble N.A., 1997) и как общий фактор, участвующий в развитии почечного интерстициального фиброза (Ito Y. et al., 1998). Исследования показали, что канальцевые эпителиальные клетки подвергаются критической трансформации по механизму эпителиально-мезенхимального перехода (Strutz F. et al., 1995). Показано, что фибробласты возникают в большом количестве локально посредством EMT (Iwano M. et al., 2002) и такие трансдифференцирующие эпителиальные клетки в почечных канальцах могут играть фундаментальную роль, приводя к возможному повреждению почек. Почечный интерстициальный фиброз и эпителиальная трансдифференцировка признаны в качестве ключевых этапов, приводящих к терминальной стадии почечной недостаточности.
ПТГрП проявляет рост-модулирующие и провоспалительные свойства в различных клеточных системах, включая тубулоэпителиальные клетки (Funk J.L., 2001; Ramila D. et al., 2008). ПТГрП сверхэкспрессируется при различных тубулоинтерстициальных нефропатиях, и его избыточная экспрессия коррелирует с развитием протеинурии как у мышей с диабетом, так и у крыс с тубулоинтерстициальным повреждением после перегрузки белком (Izquierdo A. et al., 2006; Largo R., et al., 1999). Исследование на мышах с нефротоксичностью, вызванной фолиевой кислотой, указывает на важную роль ПТГрП в развитии фиброгенеза почек (Ortega A. et al., 2006). Существуют данные указывающие на участие ПТГрП в механизмах, связанныхс прогрессированием повреждения почек. У крыс с хроническим интерстициальным фиброзом, индуцированным циклоспорином, выявлено повышение экспрессии мРНК ПТГрП почки и резкое увеличение иммуноокрашивания ПТГрП в почечной коре (Garcia-Ocana A. et al., 1998). Показано, что большее количество инфильтрирующих макрофагов было связано с повышенной пролиферацией фибробластов в интерстиции почек, поврежденных фолиевой кислотой, у мышей со сверхэкспрессией ПТГрП (Ortega A. et al., 2006). У этих мышей усиленное иммуноокрашивание на α-гладкомышечный актин ( α-SMA), маркер активированных фибробластов или миофибробластов (Strutz F. et al., 2002), также наблюдалось в почечном интерстиции после токсического поражения почек фолиевой кислотой. Эти данные, полученные в исследовании in vivo были дополнительно подтверждены результатами экспериментов in vitro, демонстрирующими, что ПТГрП (1–36) может индуцировать α-SMA и стимулировать экспрессию проколлагена и фибронектина 1-го типа в почечной фибробластной клеточной линии и в тубулоэпителиальных клетках. В совокупности эти факты свидетельствуют, что ПТГрП, по-видимому, действует как фиброгенный медиатор при токсическом повреждении почек (Ortega A. et al., 2006).
Кроме того, более интенсивное иммуноокрашивание коллагенов типов I и IV выявлено в почечном интерстиции почек у мышей с обструкцией мочеточника, сверхэкспрессирующих ПТГрП, чем у их нормальных однопометников (Ardura J.A. et al., 2008). Было обнаружено, что ПТГрП (1–36) стимулирует экспрессию обоих типов коллагенов, проколлагена 1-го типа и фибронектина в тубулоэпителиальных клетках и фибробластах почек in vitro. По крайней мере, часть этих эффектов была отменена антагонистом PTH1R (Ortega A. et al., 2006). Тубулоэпителиальные клетки также могут вносить вклад в развитие почечного фиброза путем непосредственной генерации миофибробластов посредством эпителиально-мезенхимального перехода (Ardura J.A. et al., 2008; Liu Y., 2004). Несколько исследований подтверждают важную роль различных профиброгенных факторов в процессе EMT в почках. TGF- β является наиболее известным фактором, изученным в этом отношении, и, по-видимому, включает активацию MAPKs и белков Smad. Кроме того, активация тирозинкиназы рецептора EGF (EGFR) может запускать EMT в почечных тубулоэпителиальных клетках (Gore-Hyer E. et al., 2002; Grande M. et al., 2002; Liu Y., 2004; Zhuang S. et al., 2004; Zhuang S. et al., 2005). Сообщалось, что ПТГрП стимулирует EMT посредством взаимодействия с фактором роста эндотелия сосудов (VEGF) (Ardura J.A. et al., 2008). Предполагается, что TGF- β может действовать как модулятор действия ПТГрП через PTH1R в почечных клетках. TGF- β способен опосредовать ПТГрП-индуцированную гипертрофию в висцеральных эпителиальных клетках почечных клубочков – подоцитах, где ПТГрП может индуцировать активацию TGF- β (Romero M. et al., 2010). Кроме того, стимуляция PTH1R может привести к трансактивации EGFR, что свидетельствует о важной роли ПТГрП в EMT почек. Показано, что ПТГрП способен вызывать различные фенотипические изменения, связанные с EMT в тубулоэпителиальных клетках (Ardura J.A. et al., 2010). Было обнаружено, что блокада TGF- β уменьшает фиброз почек как на экспериментальных моделях повреждения почек, так и на культивируемых клетках почек. Это позволило предположить, что TGF- β действует как нижестоящий медиатор ПТГрП. Такое же взаимодействие между этими двумя факторами наблюдалось при гипертрофии подоцитов, вызванной ПТГрП. Обнаружено, что два важных медиатора EMT, такие как белки TGF- β и EGFR, были повышены у животных с моделированием воспаления почек путем создания обструкции мочеточника, что связано с направленной сверхэкспрессией ПТГрП в проксимальных канальцах почек.
В недавнем исследовании (Chen H-M. et al., 2019) впервые была продемонстрирована вовлеченность ПТГрП в развитие редкого аутосомно-доминантного заболевания почек – фибронектиновой гломерулопатии, характеризующегося прогрессирующим массивным внутригломерулярным отложением фибронектина и нефротической протеинурией. Assmann et al., (1995) впервые показали наличие обширных отложений в мезангии и субэндотелиальном пространстве с сильной иммунной реактивностью к фибронектину. Они назвали болезнь «семейный гломерулонефрит с отложенияем фибронектина». Castelletti et al. (2008) впервые сообщили о связи между фибронектиновой гломерулопатией и тремя гетерозиготными миссенс-мутациями в гепарин-связывающих доменах гена фибронектина 1 (p.Tyr973Cys, p.Trp1925Arg и p.Leu1974). В 2016 году Ohtsubo et al. (2016) идентифицировали шесть мутаций гена фибронектина 1 и впервые сообщили о пяти мутациях (p.Pro969Leu, p.Pro1472del, p.Trp1925Cys, p.Lys1953_Ile1961del и p.Leu1974Pro) в пределах домена, связывающего интегрин. В настоящее время не существует известных эффективных и специфических методов лечения этого патологического состояния. Chen H-M. et al., (2019) показали, что воздействие ПТГрП (1–34), вызывает ROS-зависимую трансактивацию рецептора эпидермального фактора роста (EGFR), опосредованную Src-киназой, и последующую активацию Akt и ERK1/2, что приводит к увеличению экспрессии фибронектина. Продуцируемые НАДФН-оксидазой 1 активные формы кислорода опосредуют Src-зависимую трансактивацию EGFR и активацию PI3K, индуцированную ПТГрП (1–34) в мезангиальных клетках крыс. Как нижестоящие эффекторы PI3K, Akt и ERK1/2 дискретно приводят к избыточному синтезу белка фибронектина. Показано, что ПТГрП (1–34) пептид-индуцированная активация фибронектина в мезангиальных клетках крыс может быть независимой от передачи сигналов TGF- β/Smad в MC крыс. Существующие данные свидетельствуют о том, что ПТГрП, TGF- β, VEGF, а также активация рецептора эпидермального фактора роста могут действовать совместно посредством активации ERK, модулируя EMT в почечных тубулоэпителиальных клетках (Ardura J.A. et al., 2010). В совокупности эти данные демонстрируют важную роль ПТГрП в фиброгенезе почек благодаря способности этого протеина индуцировать экспрессию белков внеклеточного матрикса, а также путем модуляции EMT в почечных тубулоэпителиальных клетках.