
Научная электронная библиотека
Монографии, изданные в издательстве Российской Академии Естествознания

Попков В. М., Чеснокова Н. П., Ледванов М. Ю.,
1.2. Молекулярно-клеточные механизмы цитотоксического действия гипоксии. Патогенез гипоксического некробиоза
Как известно, динамика формирования структурных и функциональных сдвигов в различных органах и тканях при гипоксии определяется в значительной мере темпами ее развития, локализацией патологии, характером этиологических факторов, инициирующих гипоксию, и особенностями компенсаторно-приспособительных реакций в том или ином органе [36, 49].
В соответствии с данными литературы устойчивость тканей различных органов и систем к гипоксии широко варьирует. Наиболее чувствительной к гипоксии является нервная система: при полном прекращении кровотока признаки повреждения коры головного мозга обнаруживаются через несколько секунд. Снижение потребления кислорода на 20 % структурами головного мозга вызывает потерю сознания. Через 5–6 мин аноксии головного мозга возникают глубокие структурные изменения нейронов, а в продолговатом мозге – через 10–15 мин [6].
В сердечной мышце мелкие очаги некроза появляются через 3–5 мин. с момента развития ишемии, а крупноочаговый инфаркт миокарда формируется уже спустя 20–30 мин [31].
Недостаток кислорода в тканях приводит, прежде всего, к дефициту макроэргических соединений, образуемых в сопряженных с окислительно-восстановительными процессами реакциях фосфорилирования на внутренней мембране митохондрий [9, 10, 22, 37].
Основным энергетическим субстратом для нервной системы, а также для клеток других органов и тканей, является глюкоза. Между тем, при нормальной оксигенации миокарда основным источником его энергетического обеспечения являются высшие жирные кислоты. Так, при окислении 1 молекулы пальмитиновой кислоты образуется 130 М АТФ. В условиях ишемии миокарда усиливается конкурентное ингибирование использования жирных кислот лактатом, что приводит к значительному снижению энергообеспечения миокарда. Так, в процессе анаэробных гликолитических реакций энергетический выход на 1 молекулу глюкозы составляет 2 М АТФ [5, 36].
В то же время известно, что на каждую молекулу глюкозы, претерпевающую полное окисление до СО2 и воды в миокарде, печени, почках, т.е. в органах, где функционирует малат-аспартатная челночная система, образуется максимум 38 М АТФ [22].
Вышеизложенное свидетельствует о том, что независимо от характера этиологических факторов и механизмов развития гипоксии наиболее ранними проявлениями нарушения оксигенации тканей являются сдвиги их энергетического обеспечения и связанные с ними нарушения углеводного, жирового и белкового метаболизма.
Одним из метаболических признаков гипоксии и, соответственно, недостаточности энергообеспечения нервной ткани, а также миокарда является снижение уровня креатинфосфата (КФ), выполняющего роль не только резервного источника макроэргических фосфатных связей, но и обеспечивающего их транспорт в клетках к местам энергетических трат [33]. Так, уже через несколько секунд мозговая ткань теряет около 70 % КФ, а через 40–45 с. КФ полностью исчезает [11]. Почти одновременно падает уровень АТФ, увеличивается концентрация продуктов распада, так называемых метаболитов изнашивания – АДФ, АМФ, НФ, что приводит к увеличению потенциала фосфорилирования, предоставляющего собой отношение:
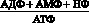 .
.
Как известно, процессы ресинтеза АТФ в митохондриях тесно связаны не только с окислительно-восстановительными реакциями, но и с реакциями гликолиза, липолиза, протеолиза, являющимися поставщиками Ац-СоА для цикла Кребса. Установлено, что регулирующими ферментами гликолиза являются фосфорилаза, гексокиназа, фосфофруктокиназа, пируваткиназа, поэтому их подавление в условиях гипоксии приводит к уменьшению образования свободной энергии и в ряде случаев носит необратимый характер. В то же время роль главного регуляторного фермента в последовательных реакциях гликолиза играет фосфофруктокиназа, которую ингибируют АТФ и цитрат, а стимулируют АМФ и АДФ [5, 22, 36].
Скорость гликолиза в условиях нормы согласована со скоростью функционирования цикла лимонной кислоты: ни пируват, ни лактат, ни ацетил-СоА обычно не накапливаются в клетках при нормальной оксигенации тканей. Согласованность между скоростью гликолиза и метаболизмом субстратов в цикле Кребса объясняется тем, что АТФ и НАД-Н являются общими компонентами для тех и других реакций. В то же время высокие концентрации АТФ и НАД-Н ингибируют реакции гликолиза. Продукт первой стадии цикла лимонной кислоты – цитрат – является аллостерическим ингибитором ключевого фермента гликолиза – фосфофруктокиназы [5, 13, 22, 46].
Таким образом, в условиях гипоксии, в случаях увеличения потенциала фосфорилирования, возникают активация ключевого фермента гликолиза – фосфофруктокиназы (ФФК) и, соответственно, возрастание пропускной способности реакции анаэробного гликолиза. При этом резко снижается запас гликогена в сердце, мозге, печени, почках, мышцах и других тканях и накапливаются продукты гликолитических реакций – молочная и пировиноградная кислоты [5, 22, 31].
Касаясь значения активации ключевого фермента гликолиза – ФФК в условиях гипоксии, необходимо отметить достаточно быструю трансформацию реакций адаптации в реакции дезадаптации, реализуемых при участии этого фермента [14].
Так, активация ФФК на начальных этапах ишемического или гипоксического повреждения клеток приводит к усилению мобилизации гликогена, несколько улучшает энергообеспечение тканей. При этом истощаются запасы гликогена, усиливается ацидоз, приводящий на пике своего развития к подавлению ФФК и полной блокаде энергообеспечения клетки [5, 36].
Развитие метаболического ацидоза при гипоксических состояниях усугубляется также недостаточностью реакций окисления жирных кислот, аминокислот, чрезмерным накоплением кислых продуктов метаболизма указанных соединений [13, 38, 50].
Что касается окисления жирных кислот в митохондриях и их роли в энергетическом обеспечении тканей, в частности, миокарда, следует отметить две главных стадии. На первой стадии происходит последовательное отщепление двууглеродных фрагментов (в виде ацетил-СоА) от карбоксильного конца цепи жирной кислоты в результате цикла ферментативных реакций. При завершении таких 7 циклов в превращениях 16 – углеродной цепи пальмитиновой кислоты образуется 8 двууглеродных фрагментов в форме ацетил-СоА. На второй стадии окисления жирных кислот ацетильные остатки ацетил-СоА окисляются через цикл лимонной кислоты до СО2 и воды в митохондриях [5, 22, 30].
На обеих стадиях окисления жирных кислот атомы водорода или соответствующие им электроны передаются по митохондриальной цепи переноса электронов на кислород. С этим потоком электронов сопряжен процесс окислительного фосфорилирования АДФ до АТФ. Следовательно, в условиях гипоксии различного генеза блокируются процессы окисления жирных кислот в тканях, в избытке накапливаются кислые продукты, формируется метаболический ацидоз и развиваются дефицит АТФ, подавление всех энергозависимых реакций [5, 22, 30, 50].
Большую часть метаболической энергии, вырабатываемой в тканях, поставляют процессы окисления углеводов и триацилглицеридов (в среднем 90 % всей энергии). Лишь 10–15 % энергии поставляется в процессе окисления аминокислот. Если аминокислоты, высвобождающиеся при обычном динамическом обновлении белков не используются для синтеза новых белков, то они подвергаются окислительному расщеплению. В случаях нарушения утилизации глюкозы возникает усиление катаболизма белков, при этом аминокислоты теряют свои аминогруппы, превращаются в ?-кетокислоты. Последние в условиях нормальной оксигенации тканей вовлекаются в цикл Кребса с образованием СО2 и воды. Естественно, что в условиях гипоксии, когда нарушаются окислительно-восстановительные реакции в цикле Кребса, развитие метаболического ацидоза усугубляется и за счет избыточного накопления в тканях аминокислот, ?-кетокислот [5, 22, 30].
Касаясь функциональной значимости метаболического ацидоза, закономерно развивающегося при гипоксии различного генеза, следует отметить ряд последующих неспецифических метаболических и функциональных расстройств, представляющих собой динамическую трансформацию реакций адаптации в реакции дезадаптации.
Как известно, типовой реакцией тучных клеток и тромбоцитов на развитие гипоксии и ацидоза является их дегрануляция с избыточным освобождением в окружающую среду высокоактивных соединений – гистамина, серотонина, ФАТ, ФХЭ, ФХН, лейкотриенов, интерлейкинов [1, 16, 51, 52]. В свою очередь, избыточное накопление ионов водорода, биологически активных соединений приводит к резкому увеличению проницаемости биологических мембран за счет структурных переходов в белках и липидах, и активации процессов свободно-радикального окисления [8, 39, 40, 46].
Таким образом, среди механизмов, приводящих к повреждению биологических мембран при гипоксии различного генеза, необходимо выделить следующие:
1) развитие метаболического ацидоза,
2) выброс вазоактивных соединений тучными клетками,
3) активацию процессов липопероксидации,
4) высвобождение лизосомальных гидролаз при дезорганизации лизосомальных мембран с последующим усугублением метаболических сдвигов.
Очевидно, что развитие гипоксического некробиоза связано в значительной мере с дезорганизацией цитоплазматических, лизосомальных, митохондриальных и др. биологических внутриклеточных мембран, формирующих отдельные функциональные и структурные компартменты.
Наиболее ранние расстройства возникают у градиентсоздающих и сократительных систем клеток.
Как известно, одним из наиболее энергоемких ферментов является Na-, К-АТФ-аза, обеспечивающая трансмембранный перенос ионов против градиента концентрации и поддерживающая таким образом уровень потенциала покоя клетки и ее возбуждение. Развитие гипоксического состояния, дефицит макроэргов, увеличение пассивной проницаемости цитоплазматических мембран клеток при их дезорганизации в условиях гипоксии приводят к развитию вначале частичной, а затем стойкой деполяризации клеток, невозможности их реполяризации и, соответственно, к отсутствию формирования потенциала действия, подавлению функциональной активности клеток. Одним из последствий подавления Na-,К-АТФ-азы и дезорганизации структурных компонентов цитоплазматических мембран, белков и липидов является избыточное проникновение в цитоплазму Na+ и Н2О с последующей гипергидратацией, развитием отека и «мутного набухания» клетки. Внутриклеточные гипергидратации – один их типичных признаков ранней обратимой стадии некробиоза клеток при гипоксии [5, 22, 30, 36, 50].
Важнейшим фактором повреждения клеток при гипоксии являются ионы кальция. Внутриклеточная концентрация кальция в состоянии покоя поддерживается в среднем на уровне 10–7 М, что в 100.000 раз меньше, чем в межклеточной жидкости. В период возбуждения кальций проникает из внеклеточной среды в клетку через потенциалзависимые кальциевые каналы. При этом возникают активация фосфолипазы С и образование липидных внутриклеточных посредников – диацилглицерина и инозинфосфамина. Цитоплазматический кальций взаимодействует с кальмодулином – внутриклеточным рецептором с последующей активацией кальмодулинзависимых протеинкиназ и включением тех или иных внутриклеточных реакций [31].
В условиях гипоксии, дефицита энергетического обеспечения клеток возникают недостаточность механизмов инактивации цитоплазматического кальция и удаления его из клеток в связи с подавлением активности АТФ-зависимого Са-насоса, натрий-кальциевого обменного механизма, дестабилизацией митохондриальных мембран и мембран эндоплазматического ретикулума, играющих в условиях нормы важную роль в поддержании баланса внутриклеточного кальция. При избытке внутриклеточного кальция усугубляются процессы набухания митохондрий, усиливается дефицит АТФ и подавление всех энергозависимых реакций в клетке [4, 26]. Избыток кальция активизирует ядерные эндонуклеазы, фрагментирующие ДНК, индуцирует апоптоз. При высоком уровне внутриклеточного кальция активизируются нейтральные протеазы – кальципаины, разрушающие цитоскелет клетки, в частности, белки фоурин и В-актин, лизирующие рецепторы и протеинкиназу С [13, 26, 31].
При гипоксическом некробиозе вокруг гибнущих клеток формируется кальцийзависимая активация системы комплемента, активация коагуляционного и тромбоцитарного звеньев гемостаза, а также фибринолиза и калликреин-кининовой системы [13, 50].
Активация под влиянием кальция мембранных фосфолипаз приводит к дальнейшей дезинтеграции мембран клеток, активации циклооксигеназы и липооксигеназы с последующим образованием простагландинов, лейкотриенов, свободных радикалов с выраженным цитотоксическим действием [27, 45, 52].
Чрезвычайно важна роль дезинтеграции митохондриальных мембран в механизмах гипоксического некробиоза клеток [40].
Как известно, в клетках эукариот все специфические дегидрогеназы принимают участие в окислении пирувата и других субстратов, локализованных в митохондриальном матриксе. Во внутренней мембране митохондрий локализуются переносчики электронов, составляющие дыхательную цепь и ферменты, катализирующие синтез АТФ из АДФ и фосфата [22, 36].
В связи с этим очевидно, что продукты гликолиза, липолиза, протеолиза, вовлекаемые через ацетил-СоА в цикл Кребса, а также АДФ должны пройти через обе митохондриальные мембраны, в то время как новообразованные АТФ проникают из внутренней мембраны митохондрий в цитоплазму клетки и далее к местам энергетических трат. Установлено, что наружная мембрана легко проницаема для всех молекул и ионов небольшого размера, в то время как во внутренней мембране имеются специальные ферментативные транспортные системы, обеспечивающие трансмембранный перенос ионов и различных соединений [39, 40].
Согласно хемиосматической гипотезе функция переноса электронов, происходящего на внутренней митохондриальной мембране, заключается в том, чтобы откачивать ионы Н+ из матрикса митохондрий в наружную среду для создания градиента концентрации ионов Н+ между двумя водными фазами, разделяемыми внутренней мембраной митохондрий, и накопления потенциальной энергии. Очевидно, что при нарушении целостности структуры митохондриальной мембраны в условиях гипоксии возникает утечка ионов Н+ через мембраны, в процессе которой не исключается возможность образования активных форм кислорода с одной стороны, и недостаточность ресинтеза АТФ – с другой [5, 22, 36, 39, 40].
Таким образом, при избыточном накоплении ионов кальция в клетке, активации процессов липопероксидации при гипоксии различного генеза резко повышается проницаемость митохондриальных мембран, возникает набухание митохондрий, пространственная дезориентация ферментативных систем транспорта электронов, синтеза АТФ. В результате происходят разобщение окислительного фосфорилирования и дыхания и подавление всех энергозависимых систем клетки: синтеза белка, трансмембранного переноса ионов, сопряжения процессов возбуждения и сокращения в мышечных структурах и т.д. [5, 13, 22, 36].
В процессе набухания митохондрий энергия потока электронов трансформируется в тепловую энергию.
Наряду с локальными и системными метаболическими сдвигами в тканях, обусловленными гипоксией, ацидозом, активизацией процессов липопероксидации при гипоксии различного генеза возникает комплекс метаболических и функциональных сдвигов, обусловленных выбросом гормонов адаптации – катехоламинов, глюкокортикоидов.
Как известно, при чрезмерной активации симпатоадреналовой системы (САС) реакции адаптации довольно быстро трансформируются в дезадаптационные процессы [12]. Во-первых, при активации освобождения норадреналина происходит спазм сосудов периферических органов и тканей и, соответственно, усугубление циркуляторной гипоксии. На фоне активации САС при участии постсинаптических ?-адренорецепторов возможна активация процессов гликолиза, гликогенолиза, липолиза, что, безусловно, усугубляет развитие ацидотических сдвигов, свойственных гипоксии [14].
Усиление адренергических влияний закономерно сопровождается активацией процессов липопероксидации, что вносит весомый вклад в механизмы развития гипоксического некробиоза клеток органов и тканей, чувствительных к ишемии.
Синхронно с освобождением катехоламинов в условиях гипоксического стресса выбрасываются глюкокортикоиды, индуцирующие процессы лизиса и апоптоза в лимфоидной ткани, блокирующие процессы пролиферации и репаративной регенерации в ряде внутренних органов.