
Научная электронная библиотека
Монографии, изданные в издательстве Российской Академии Естествознания

ЦИТОГЕНЕТИЧЕСКИЕ, МОЛЕКУЛЯРНЫЕ И КЛИНИЧЕСКИЕ ОСНОВЫ ГЕНЕТИЧЕСКИ ОБУСЛОВЛЕННЫХ БОЛЕЗНЕЙ
Юров И. Ю., Ворсанова С. Г., Воинова В. Ю., Чурносов М. И., Юров Ю. Б.,
5.3. Митохондриальное наследование
Митохондриальное (цитоплазматическое) наследование характерно для особого класса наследственной патологии – митохондриальных болезней. Каждая клетка содержит тысячи копий митохондриальной ДНК. Ряд редких болезней с необычной комбинацией неврологических и миопатических признаков, кардиомиопатии, диабет, как оказалось, возникают вследствие мутаций митохондриальных генов. Неудивительно, что головной мозг, мышцы и сердце поражаются в большей степени, поскольку эти органы наиболее энергозависимы. Митохондриальные болезни поражают оба пола, но передаются только через женщин (рис. 15). У большинства людей митохондриальная ДНК идентична во всех митохондриях (гомоплазмия). В случае мутаций ДНК в части митохондрий, у индивидуума будет две популяции митохондрий – нормальные и мутантные, т. е. гетероплазмия. Доля митохондрий с мутантной ДНК варьирует между клетками и тканями. Это является объяснением различной тяжести течения заболевания у людей с митохондриальными болезнями. Ряд митохондриальных белков кодируется ядерными генами, а мутации в них нарушают функцию митохондрий. Напрмер, мутации генов белков комплекса цитохрома С наследуются аутосомно-рецессивно, а мутации Х-сцепленного гена G4.5 (TAZ) вызывают синдром Барта (кардиоскелетную миопатию с нейтропенией и аномальными митохондриями) у мальчиков.
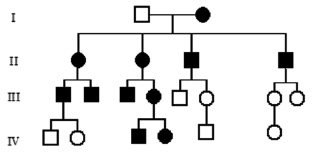
Рис. 15. Родословная при митохондриальном типе наследования
Множественные аллели и комплексные признаки
Выше рассмотрены признаки, с которыми связаны только два аллеля – нормальный и мутантный. Некоторые гены имеют более двух аллельных форм, т. е. множественные аллели. Некоторые из них могут быть доминантными, другие – рецессивными по отношению к нормальному аллелю. Пример множественных аллелей – наследование групп крови человека.
Развитие генетики сделало возможным исследование комплексных признаков, которые формируются при взаимодействии нескольких генов. На этой основе возникла концепция олигогенного (дигенного и триаллельного) наследования.
При дигенном наследовании наблюдается аддитивный эффект гетерозиготных мутаций в двух различных локусах. Например, одна из форм пигментного ретинита, приводящая к потере зрения, вызвана гетерозиготностью по мутациям двух генов (ROM1 и PRPH). Оба эти гена кодируют белки, присутствующие в фоторецепторах сетчатки глаза. Индивидуумы, гетерозиготные по мутации только одного из этих двух генов, не имеют клинических проявлений.
Триаллельное наследование можно рассмотреть на примере синдрома Барде-Бидля – редкого заболевания, характеризующегося ожирением, полидактилией, аномалиями почек, пигментным ретинитом и когнитивными нарушениями. Семь различных генных локусов, мутации в которых ведут к синдрому Барде–Бидля, были идентифицированы. До недавнего времени считалось, что заболевание наследуется аутосомно-рецессивно. Однако, сейчас известно, что есть одна форма синдрома, когда индивидуум, гомозиготный по мутациям одного локуса, является также гетерозиготным по мутации другого локуса. Таким образом, для того, чтобы заболевание проявлялось, необходимо три мутантных аллеля.
Антиципация. При некоторых аутосомно-доминантных болезнях манифестация симптомов более ранняя и течение болезни более тяжелое у потомков по сравнению с их родителями, также страдающими этим заболеванием. Феномен увеличения тяжести болезни из поколения в поколение называют антиципацией. Одним из объяснений антиципации является экспансия нестабильных триплетных повторов. В качестве примеров можно привести такие болезни экспансии триплетных повторов, как миотоническая дистрофия, хорея Гентингтона, болезнь Кеннеди.