
Научная электронная библиотека
Монографии, изданные в издательстве Российской Академии Естествознания

Генеалогический анализ семей с Х-сцепленной умственной отсталостью
Нами проведен анализ 595 родословных детей с Х-сцепленной умственной отсталостью, включавших сведения о 19 789 индивидуумах (табл. 13).
Таблица 13
Результаты генеалогического анализа в семьях с X-сцепленной умственной отсталостью (N = 595)
|
Нозологические формы |
Число проанализированных родословных |
Число семейных случаев |
Удельный вес семейных случаев среди всех проанализированных |
|
Синдромы, проявляющиеся преимущественно у девочек-гетерозигот, в том числе: |
369 |
2 |
0,5 % |
|
Блоха-Сульцбергера |
13 |
1 |
8 % |
|
RTT |
354 |
1 |
0,25 % |
|
Айкарди |
1 |
0 |
0 % |
|
Гольтца |
1 |
0 |
0 % |
|
Синдромы, проявляющиеся преимущественно у мальчиков – гемизигот, в том числе: |
208 |
35 |
17 % |
|
Синдром FRAXA |
43 |
6 |
14 % |
|
Мукополисахаридоз II типа |
18 |
1 |
6 % |
|
Редкие синдромы |
147 |
23 |
16 % |
|
Несиндромальные формы Х-сцепленной умственной отсталости |
18 |
18 |
100 % |
Как видно из табл. 13, генеалогическое исследование имело наибольшее значение в семьях больных с несиндромальной Х-сцепленной умственной отсталостью (n = 18), поскольку Х-сцепленный характер наследования нарушений интеллекта служил единственным критерием, согласно которому в этих семьях и была идентифицирована MRX. В каждой из 18 проанализированных родословных умственная отсталость наблюдалась минимум у двух индивидуумов мужского пола: в четырёх семьях – у полусибсов мужского пола по линии матери, в двух семьях – у дяди и племянника, в остальных родословных – у трех и более мужчин, принадлежащих к разным поколениям. Ниже приведены фрагменты родословных с тремя и более случаями несиндромальной Х-сцепленной умственной отсталости (рис. 19). Генеалогический анализ был малоинформативен в диагностике Х-сцепленных синдромов, проявляющихся преимущественно у гетерозигот. Только в одной из 351 семьи с RTT заболевание встречалось более чем у одного индивидуума, а в остальных было спорадическим. Согласно зарубежным данным, 99 % случаев RTT – спорадические, описано лишь несколько семейных [Trappe et al., 2001]. Единственный семейный случай RTT наблюдался нами у полусибсов по линии матери, что возможно было объяснить наличием у неё гонадного мозаицизма по MECP2-мутации. Еще в двух родословных наблюдались монозиготные близнецы с RTT. Монозиготных близнецов обычно не относят к семейным случаям, т.к. их разделение происходит после образования зиготы. Таким образом, удельный вес семейных случаев RTT в исследуемой группе был 1/391 (0,25 %), т.е. вдвое ниже, чем в наблюдаемых другими авторами когортах [Shahbazian, Zoghbi, 2001].
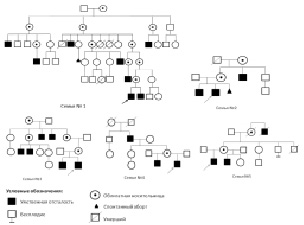
Рис. 19. Фрагменты родословных семей с несиндромальной Х-сцепленной умственной отсталостью
В проанализированных родословных девочек с синдромами Гольтца и Айкарди не было обнаружено больных с аналогичной патологией. Наши данные совпадают с литературными, согласно которым летальные для индивидуумов мужского пола Х-сцепленные доминантные синдромы (Айкарди, RTT) представляют собой, как правило, единичные случаи в родословных. Последнее, однако, не относится к синдрому Блоха-Сульцбергера. В одной из 13 родословных девочек с синдромом Блоха-Сульцбергера заболевание наблюдалось у матери и дочери. Ещё у одной из матерей было два спонтанных аборта на ранних сроках беременности, что позволяло предполагать носительство ею заболевания, а спонтанные аборты расценивать как возможную элиминацию плодов мужского пола. Авторами, исследовавшими значительное число семей с синдромом Блоха-Сульцбергера, было показано, что повторные его случаи в родословных нередки [Parrish et al., 1996]. Доля случаев заболеваний, которые являются результатом мутаций de novo, согласно данным литературы, составляет по 99,5 % для синдромов Айкарди и RTT, 90 % – для синдрома Гольтца и 65 % – для синдрома Блоха-Сульцбергера [Hagberg et al., 2002; Fusco et al., 2004].
Генеалогический анализ сыграл значимую роль в диагностике синдромальных форм Х-сцепленной умственной отсталости, проявляющихся преимущественно у гемизигот: в 35 из 208 (17 %) проанализированных родословных заболевание наблюдалось более чем у одного мальчика, т.е. получены доказательства сцепленного с хромосомой Х наследования заболевания (синдромы умственной отсталости, сцепленной с ломкой хромосомой Х, FG, Менкеса, Аарскога и др.).
Наибольший интерес представляли родословные детей с синдромом умственной отсталости, сцепленной с ломкой хромосомой Х. Синдром известен своим необычным типом наследования с увеличением пенетрантности мутантного гена при передаче из поколения в поколение (парадокс Шермана) [Sherman et al., 1985]. Генеалогический анализ позволил установить среди 43 проанализированных родословных 6 семейных случаев заболевания: у родных брата и сестры (1 семья), у мальчика и его двоюродной сестры по линии матери (1 семья), у родных братьев (2 семьи), у пробанда и его дяди по линии матери (1 семья) и у двух братьев и сестры (1 семья). Кроме того, у ряда родственников отмечались фенотипические признаки, связанные с экспансией тринуклеотидных повторов в промоторе гена FMR1. Так, у 7 матерей больных мальчиков с синдромом FRAXA наблюдались трудности в обучении, у четырёх – снижение памяти, у одной матери был пограничный интеллект. Несколько матерей с сохранным интеллектом имели такие психологические особенности, как тревожные состояния (5), социофобии (2). В родословных детей с синдромом FRAXA прослеживались случаи синдрома преждевременного нарушения функции яичников. Так, у 13 женщин (6 матерей, одной тети, 4 бабушек, 1 двоюродной бабушки и 1 прабабушки) наблюдалось прекращение менструаций в возрасте до 40 лет. Кроме того, у ряда женщин отмечались заболевания женских половых органов: дисфункция яичников (7 матерей), миомы матки (4 матери). Одна из матерей умерла от рака шейки матки в 38 лет, бабушка по материнской линии в этой же семье оперирована по поводу опухоли яичников. У 6 мужчин (4 деда и 2 прадеда по линии матери) наблюдался синдром тремора и атаксии, отмечен тремор головы при волнении у одной из матерей с возраста 43-х лет, что указывало на развитие у неё данного синдрома. У 5 матерей и 4-х сестер в семьях отмечены характерные для данного синдрома лицевые микроаномалии (крупные ушные раковины, выступающий лоб и подбородок). Пример родословной наблюдавшейся нами семьи с синдромом FRAXA представлен на рис. 20.

Рис. 20. Родословная семьи, в которой наблюдались сразу три фенотипа, ассоциированных с экспансией тринуклеотидных повторов в промоторе
гена FMR1: синдром умственной отсталости, сцепленной с ломкой хромосомой Х, наблюдался у пробанда и его дяди по линии матери, у тети и бабушки по линии матери – преждевременное нарушение функции яичников, у прадеда – синдром тремора и атаксии
Таким образом, генеалогический анализ в семьях с синдромом FRAXA помимо Х-сцепленного типа наследования показал, что практически в каждой родословной наблюдалось от одного до нескольких индивидуумов, у которых проявлялись симптомы, связанные с полной мутацией или премутацией гена FMR1. Это указывало на необходимость исследований числа CGG- повторов в промоторе гена FMR1 у значительного числа индивидуумов в каждой семье в целях проведения корректного медико-генетического консультирования.
Клинико-генеалогический анализ в семьях с различными редкими синдромальными формами XLMR у мальчиков позволил выявить среди 147 проанализированных родословных 23, в которых было более одного больного ребенка. В этих родословных наблюдалось Х-сцепленное наследование заболеваний. Пример таких родословных представлен на рис. 21. На рис. 21, А представлен фрагмент родословной семьи, в которой синдром FG наблюдался у двух родных братьев. Обследование их дяди по линии матери показало, что он был низкого роста, имел легкую умственную отсталость, относительную макроцефалию, хронические запоры. Выявленный симптомокомплекс позволил c высокой вероятностью предполагать у этого родственника синдром FG. Мать больных сибсов была низкого роста и с девятилетнего возраста страдала эпилепсией, что указывало на гетерозиготное носительство ею данного синдрома. В семье, фрагмент родословной которой представлен на рис. 21, Б, у матери ребенка с синдромом Менкеса был сын от первого брака, который умер в возрасте 7 месяцев. Этот мальчик имел клинические проявления, аналогичные наблюдавшимся у пробанда: глубокую задержку развития, судороги и светлые тонкие курчавые волосы (pili torti). Это позволило предположительно рассматривать случай синдрома Менкеса как семейный и считать мать облигатной носительницей заболевания.
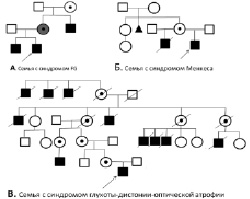
Рис. 21. Родословные семей с редкими Х-сцепленными синдромами
В родословной пробанда с синдромом глухоты-дистонии-оптической атрофии (синдромом Мора-Транебьерг), представленной на рис. 21, B, наблюдалось большое число родственников мужского пола с одинаковой патологией. У всех больных отмечалась потеря зрения, слуха, двигательные нарушения и деменция. В данной семье анализ родословной помог диагностике синдрома глухоты-дистонии-оптической атрофии у пробанда. Благодаря генеалогическому анализу, диагностический поиск был сужен до группы Х-сцепленных форм умственной отсталости. В опубликованных рядом авторов наблюдениях синдрома глухоты-дистонии-оптической атрофии, так же как и в нашей работе, отмечалось большое количество больных родственников. Так, в одной из семей обнаружено 16 больных мужчин в пяти поколениях [Tranebjærg et al., 2000].
Суммируя данные о стертых клинических проявлениях у женщин из семей с Х-сцепленными синдромами, характерными для гемизигот, следует отметить, что в настоящей работе различная пенетрантность заболевания у женщин-гетерозигот наблюдалась нами в не менее трети проанализированных родословных. Например, у носительниц Х-сцепленных мутаций наблюдались следующие клинические признаки: аномалии пальцев при ото-палато-дигитальном синдроме 1 типа, эпилепсия и низкий рост при синдроме FG; когнитивные нарушения и лицевые микроаномалии при синдроме FRAXA; конической формы пальцы, опущенные наружные углы глаз с периорбитальной полнотой тканей, полные губы при синдроме Коффина-Лоури; катаракта при синдроме Лоу; малые размеры окружности головы и когнитивные нарушения при Х-сцепленной спастической диплегии с умственной отсталостью и др. Идентификация носительниц заболеваний в остальных случаях была невозможной без применения широкого спектра лабораторных генетических методов.
Необходимо отметить, что в последние годы некоторыми исследователями критикуется традиционное разделение сцепленных с хромосомой Х заболеваний на Х-сцепленные доминантные и рецессивные и предлагается использовать понятие просто Х-сцепленного наследования [Dobyns et al., 2004; Migeon, 2007; Pinto et al., 2010]. В своём утверждении эти авторы основываются на высокой пенетрантности, наблюдавшейся у гетерозигот при тех Х-сцепленных болезнях, которые было принято считать рецессивными. Авторы подчёркивают, что концепция Х-сцепленного доминантного/рецессивного наследования была разработана на модели дрозофилы, у которой механизм дозовой компенсации для генов хромосомы Х отличается от такового у человека (в её клетках активны обе хромосомы Х). На основании наблюдавшихся нами при большинстве форм XLMR промежуточных между нормой и заболеванием фенотипов у женщин, которые не соответствуют Х-сцепленному рецессивному наследованию, принятие концепции просто Х-сцепленного наследования представляется целесообразным.