
Научная электронная библиотека
Монографии, изданные в издательстве Российской Академии Естествознания

Возможные корреляции генотипа и фенотипа при Х-сцепленных формах умственной отсталости и прогнозирование тяжести заболеваний у детей
В нашей работе проведен анализ зависимости фенотипа детей с синдромами FRAXA и RTT от генетических (изменения последовательности ДНК генов МЕСР2 и FMR1) и эпигенетических (особенности инактивации хромосомы Х) факторов.
После открытия мутаций гена МЕСР2 у больных с RTT [Amir et al., 1999] были проведены исследования, посвященные поиску связи между типом и позицией мутации гена МЕСР2 и тяжестью течения заболевания. Полученные авторами результаты носили противоречивый характер (табл. 21).
Таблица 21
Исследования корреляций генотипа (типа и позиции мутации) и фенотипа при RTT
|
Автор |
Доля детей с мутациями гена MECP2 среди |
Корреляции генотип/фенотип: |
|||
|
Тип мутации: |
Тяжесть заболевания |
||||
|
в доменах МЕСР2 |
при отдельных мутациях |
||||
|
с тяжестью течения заболевания |
с отдельными клиническими признаками |
||||
|
1 |
2 |
3 |
4 |
5 |
6 |
|
Amir et al., 2000 |
54 из 71 спорадических |
Корреляция не обнаружена |
Миссенс мутации ведут к менее тяжелой дыхательной дисфункции, но более тяжелому сколиозу |
Не исследованы |
Не исследованы |
|
Cheadle et al., 2000 |
44 из 55 классических |
Миссенс мутации ведут к менее тяжелому фенотипу по сравнению с мутациями нонсенс и со сдвигом рамки считывания |
Корреляция |
Мутации, расположенные ближе |
Крупные делеции ~500 пн по сравнению с небольшими делециями на 3′ конце гена имеют более тяжелые клинические проявления |
|
Huppke et al., 2000 |
24 из 31 девочек |
Миссенс мутации ведут к менее тяжелому фенотипу |
Корреляция |
Не исследованы |
258–259delCA ведет к тяжелому течению заболевания |
|
Hoffbuhr et al., 2001 |
101 из 160 больных с RTT |
Корреляция не обнаружена |
Микроцефалия больше выражена у детей с нонсенс мутациями и мутациями со сдвигом рамки считывания |
Мутации, расположенные ближе |
Не исследованы |
|
Schanen et al., 2004 |
85 случаев. Нет данных о количестве больных с мутациями |
Миссенс мутации ведут к менее тяжелому фенотипу |
У детей с миссенс мутациями лучше развиты речевые навыки |
Корреляция |
R306C – легкое течение болезни, позднее начало стадии регресса, сохранность речевых и двигательных навыков |
|
Colvin et al., 2004 |
129 из 196 девочек |
Корреляция не обнаружена |
Мутации со сдвигом рамки считывания ведут к более позднему началу стадии регресса, чем нонсенс мутации. |
Наибольшая тяжесть течения RTT наблюдается при мутациях, локализованных в TRD |
R168X – наиболее ранний возраст нарушений контакта, R270X – тяжелое течение болезни, R294X – легкое течение, R306C – позднее начало регресса |
|
Bebbington et al., 2008 |
Не установлена |
Не исследованы |
Не исследованы |
Не исследованы |
Тяжелое течение при R255X и R270X, легкое –R133C и R294X - |
Так, в некоторых работах утверждалось, что миссенс мутации приводят к легкому течению заболевания по сравнению с нонсенс мутациями и мутациями со сдвигом рамки считывания, а также что мутации, расположенные дистальнее, ближе к 3’– концу гена, имеют относительно легкие клинические проявления [Weaving et al., 2003; Schanen et al., 2004]. Однако в других исследованиях вышеупомянутых корреляций генотипа и фенотипа не было обнаружено [Huppke et al., 2000; Hoffbuhr et al., 2001; Colvin et al., 2004].
Несовершенство используемых ранее клинических шкал могло быть причиной противоречивости результатов анализа корреляций генотипа и фенотипа при RTT. Эти исследования до сих пор не потеряли свою актуальность, поскольку позволяют прогнозировать развитие признаков заболевания на основании знания мутации гена МЕСР2. Кроме того, в ряде случаев идентичные мутации вызывают различные клинические проявления. Например, женщина с небольшими нарушениями обучения может иметь ту же МЕСР2 мутацию, что и ее сестра с RTT. В основе фенотипических различий в таких случаях лежат особенности Х-инактивации у индивидуумов. Х-инактивация приводит к определенному соотношению популяций нейронов с нормальным и мутантным аллелями гена МЕСР2 на активной хромосоме Х. Отсутствие развернутой клинической картины RTT обусловлено «благоприятным» сдвигом Х-инактивации [Hoffbuhr et al., 2001; Zappella et al., 2001; Shahbazian, Zoghbi, 2002; Weaving et al., 2003]. Наиболее полно исследования инактивации хромосомы Х при данном заболевании представлены в работе Юрова И.Ю. с соавторами [2005 б], в которой показано, что феномен неравной инактивации хромосомы Х является характерной особенностью RTT. При этом неравная Х-инактивация является определяющим клинический полиморфизм синдрома фактором, а течение болезни зависит от направления сдвига Х-инактивации. Различия особенностей Х-инактивации не всегда объясняют фенотипическую вариабельность между индивидуумами с RTT и одной и той же мутацией. Было показано, что значительный вклад в нее вносят гены-модификаторы, связанные с MECP2 общими патогенетическими путями. Например, частый у здоровых индивидуумов полиморфизм гена BDNF (одного из генов-мишеней для MECP2) коррелирует с предрасположенностью к судорожному синдрому и с его тяжестью при RTT [Zeev et al., 2009].
Корреляции генотипа и фенотипа исследовались нами у 74 детей с RTT: 2х мальчиков и 72-х девочек. Средний возраст детей составил 47 месяцев, варьируя от 12 месяцев до 10 лет 4 месяцев. Для статистического анализа данных об экспрессивности клинических признаков у детей с RTT, полученных при помощи разработанной клинической шкалы, нами использовался тест непараметрического сравнения независимых выборок Манна – Уитни, поскольку распределение этих данных не соответствовало критериям нормального (гауссовского) распределения. Использование t-теста было возможным только для статистической обработки результатов суммарной оценки фенотипа в баллах, поскольку их распределение было гауссовским. Проведен анализ зависимости клинической формы и тяжести течения RTT от 1) типа мутации, 2) позиции мутации в данном гене, 3) особенностей инактивации хромосомы Х. Данные о мутациях, особенностях инактивации хромосомы Х, клинической форме заболевания и суммарной оценке фенотипа в баллах у детей с RTT представлены в табл. 22. Для анализа влияния типа мутации на тяжесть течения заболевания были исследованы две группы детей с мутациями гена МЕСР2. В первую группу вошли пациенты, у которых обнаружены нонсенс мутации и мутации со сдвигом рамки считывания. Поскольку оба упомянутых типа мутаций приводят к сходным последствиям для структуры белка – синтезу укороченной полипептидной цепи, дети с данными двумя типами мутаций были объединены в первую группу. Во вторую группу вошли дети с миссенс мутациями, которые ведут к замене одной из аминокислот в белке.
Таблица 22
Мутации гена MECP2, инактивация хромосомы Х и особенности фенотипа (клиническая форма заболевания и суммарная оценка фенотипа в баллах) у больных с RTT
|
№ п/п |
Возраст, месяцы |
Мутация гена МЕСР2 |
Инактивация хромосомы Х |
Форма |
Суммарная оценка фенотипа в баллах |
|
1 |
40 |
S65X |
86:14 |
Стёртая |
26 |
|
2 |
28 |
R106W |
– |
Классическая |
31 |
|
3 |
33 |
R106W |
– |
Классическая |
32 |
|
4 |
72 |
R133C |
84:16 |
Классическая |
30 |
|
5 |
102 |
R133C |
11:89 |
Стёртая |
14,5 |
|
6 |
33 |
R133C |
98:2 |
Стёртая |
19 |
|
7 |
124 |
D156fsX172 |
17:83 |
Классическая |
43 |
|
8 |
21 |
T158M |
– |
Классическая |
61 |
|
9 |
72 |
T158M |
69:31 |
Классическая |
35 |
|
10 |
51 |
T158M |
51:49 |
Классическая |
45 |
|
11 |
24 |
T158M |
69:31 |
Классическая |
38 |
|
12 |
31 |
T158M |
– |
Классическая |
37 |
|
13 |
51 |
T158M |
– |
Классическая |
36,5 |
|
14 |
32 |
T158M |
– |
Классическая |
37 |
|
15 |
24 |
T158M |
58:42 |
Классическая |
41,5 |
|
16 |
19 |
R168X |
21:79 |
Классическая |
32 |
|
17 |
53 |
R168X |
80:20 |
Классическая |
36 |
|
18 |
48 |
R168X |
90:10 |
Классическая |
46 |
|
19* |
20 |
R168X |
– |
Классическая форма у мальчика |
44,5 |
|
20 |
53 |
R168X |
75:25 |
Классическая |
38 |
|
21 |
34 |
R168X |
– |
Классическая |
65 |
|
22 |
25 |
R168X |
– |
Классическая |
49 |
|
23 |
34 |
R168X |
58:42 |
Классическая |
61 |
|
24 |
48 |
T197M |
51:49 |
RTT-подобный фенотип |
29 |
|
25 |
30 |
A202fsX209 |
– |
Классическая |
36,5 |
|
26 |
44 |
K210G |
– |
Классическая |
32 |
|
27 |
69 |
G231fsX247 |
65:35 |
Классическая |
53 |
|
28 |
34 |
G232fsX235 |
– |
Классическая |
38 |
|
29 |
32 |
G237fsX247 |
– |
Классическая |
43,5 |
|
30 |
51 |
R250fsX288 |
96:4 |
Стёртая |
18 |
|
31 |
35 |
R255X |
22:78 |
Классическая |
39 |
|
32 |
26 |
R255X |
– |
Классическая |
60 |
|
33 |
104 |
R255X |
79:21 |
Классическая |
55 |
|
34 |
45 |
R255X |
80:20 |
Классическая |
37,5 |
|
35 |
33 |
R255X |
– |
Классическая |
42,5 |
|
36 |
28 |
R255X |
– |
Классическая |
49 |
|
37 |
52 |
R255X |
– |
Классическая |
48,5 |
|
38** |
12 |
R255X |
80:20 |
Классическая |
49 |
|
39** |
12 |
R255X |
56:44 |
Классическая |
57 |
|
40 |
36 |
R255X |
60:40 |
Классическая |
42 |
|
41 |
39 |
Q262X |
66:34 |
Классическая |
38 |
|
42 |
21 |
G269fsX288 |
47:53 |
Классическая |
56 |
|
43 |
33 |
G269fsX288 |
– |
Классическая |
36 |
|
44* |
27 |
R270X |
– |
Классическая форма у мальчика |
57 |
|
45 |
96 |
R270X |
44:56 |
Классическая |
47 |
|
46 |
78 |
R270X |
– |
Классическая |
67 |
|
47 |
22 |
R270X |
71:29 |
Классическая |
31,5 |
|
48 |
18 |
R270X |
55:45 |
Классическая |
66 |
|
49 |
46 |
R270X |
– |
Классическая |
51 |
|
50 |
36 |
R270X |
– |
Классическая |
47,5 |
|
51 |
12 |
R294X |
51:49 |
Классическая |
38 |
|
52 |
108 |
R294X |
88:12 |
Стёртая |
27 |
|
53 |
94 |
P302L |
– |
Классическая |
32 |
|
54 |
108 |
R306C |
76:24 |
Классическая |
30,5 |
|
55 |
41 |
R306C |
– |
Классическая |
31,5 |
|
56 |
96 |
R306C |
90:10 |
Форма с сохранной речью |
25 |
|
57 |
36 |
R306C |
– |
Классическая |
35 |
|
58 |
41 |
R306C |
– |
Классическая |
32 |
|
59 |
118 |
R306C |
6:94 |
Форма с сохранной речью |
23,5 |
|
60 |
47 |
R306C |
73:27 |
Стёртая |
11 |
|
61 |
34 |
356del92 |
60:40 |
Стёртая |
21 |
|
62 |
84 |
S357fsX387 |
– |
Классическая |
38 |
|
62 |
26 |
S360fsX388 |
– |
Классическая |
36 |
|
63 |
31 |
386del15 |
– |
Классическая |
33 |
|
64 |
54 |
L386fsX395 |
46:54 |
Классическая |
31,5 |
|
65 |
108 |
P388T |
95:5 |
Классическая |
40 |
|
66 |
66 |
P388fsX399 |
– |
Классическая |
38 |
|
67 |
15 |
– |
– |
Форма с ранним началом судорог |
60 |
|
68 |
46 |
– |
– |
Классическая |
39 |
|
69 |
38 |
– |
– |
Классическая |
38 |
|
70 |
20 |
– |
– |
Врожденная форма |
41,5 |
|
71 |
102 |
– |
– |
Стёртая |
13 |
|
72 |
42 |
– |
– |
Классическая |
56 |
|
73 |
39 |
– |
– |
Классическая |
30 |
|
74 |
55 |
– |
– |
Врожденная форма |
61,5 |
Примечания: * – мальчики с классической формой RTT;
** – конкордантные близнецы с RTT.
Для каждого отдельно взятого симптома RTT, оцененного по предлагаемой шкале, была обнаружена следующая закономерность: среди больных с максимальной экспрессивностью признака преобладали дети с нонсенс мутациями и мутациями со сдвигом рамки считывания, в то время как среди больных с низкой экспрессивностью признака чаще встречались дети с миссенс мутациями. Например, у больных с тяжелой микроцефалией (80 %) обнаруживались нонсенс мутации и мутации со сдвигом рамки считывания (80 %), и, напротив, у детей без микроцефалии обнаружены миссенс мутации (63 %) гена МЕСР2. Все дети, у которых наблюдались частично сохранные речевые навыки, включая отдельные слова и фразы из двух слов, имели миссенс мутации, в отличие от больных с полным отсутствием речи, у 100 % которых были обнаружены нонсенс мутации и мутации со сдвигом рамки считывания (рис. 38). В первой группе больных (с нонсенс мутациями и мутациями со сдвигом рамки считывания) суммарная оценка фенотипа в баллах (44,6 балла), следовательно, течение заболевания было статистически достоверно тяжелее по сравнению с детьми второй группы с миссенс мутациями (33,8 баллов, t = 4,031, p < 0,001). Делеции, расположенные на 3′ конце гена МЕСР2, составляют особую группу мутаций, которые не затрагивают основных функциональных доменов белка МЕСР2 и обычно приводят к клинически легким формам RTT, хотя формально они в большинстве своем являются мутациями со сдвигом рамки считывания. Поэтому, чтобы избежать противоречия, случаи делеций на 3′-конце гена были исключены из анализа влияния типа мутации на фенотип (табл. 23).
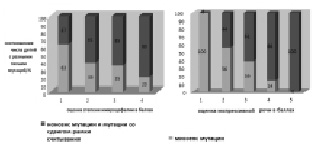
Рис. 38. Соотношения количества детей с нонсенс мутациями и мутациями со сдвигом рамки считывания (1-й группы) и миссенс мутациями (2-й группы) в зависимости от экспрессивности клинических признаков: микроцефалия, нарушения экспрессивной речи
Как видно из табл. 23, проведенный анализ продемонстрировал достоверные различия между группами индивидуумов с миссенс мутациями и больных с нонсенс мутациями и мутациями со сдвигом рамки считывания (за исключением делеций на 3′-конце гена)
по экспрессивности четырнадцати клинических симптомов и суммарной оценке фенотипа. Обнаружены статистически значимые различия между группами по таким признакам, как нарушение контакта (р < 0,05), нарушение эмоционального общения (р < 0,05), расстройства понимания обращенной речи (р < 0,05) и экспрессивной речи (р < 0,01), нарушение внимания (р < 0,05), нарушение вертикализации (р < 0,05), ходьбы (р < 0,05), преодоления препятствий (р < 0,05), расстройства вегетативных функций (р < 0,05), оро-моторных функций (р < 0,01), апраксия движений рук (р < 0,01), стереотипии (р < 0,05), спастичность мышц конечностей (р < 0,05), бруксизм (р < 0,05), сколиоз (р < 0,05).
Таблица 23
Сравнение экспрессивности клинических признаков у детей с миссенс мутациями (N = 24) и детей с нонсенс мутациями, мутациями со сдвигом рамки считывания за исключением делеций на 3′-конце гена MECP2 (N = 36) c использованием критерия Манна-Уитни для двух независимых выборок
|
Клинический признак |
Количество детей, |
p |
|||||||||||
|
Дети с миссенс мутациями (N = 24) |
Дети с нонсенс мутациями и мутациями со сдвигом рамки считывания (N = 36) |
||||||||||||
|
0 |
1 |
2 |
3 |
4 |
5 |
0 |
1 |
2 |
3 |
4 |
5 |
||
|
контакт |
1 |
12 |
10 |
1 |
0 |
12 |
20 |
3 |
0,021* |
||||
|
эмоциональное общение |
10 |
9 |
4 |
1 |
1 |
15 |
12 |
8 |
0,022* |
||||
|
понимание обращенной речи |
1 |
11 |
8 |
4 |
0 |
19 |
21 |
6 |
0,042* |
||||
|
экспрессивная речь |
0 |
1 |
6 |
11 |
2 |
1 |
0 |
0 |
4 |
12 |
18 |
2 |
0,006** |
|
внимание |
1 |
12 |
12 |
1 |
2 |
14 |
17 |
3 |
0,034* |
||||
|
Предметно-игровая деятельность |
5 |
6 |
11 |
2 |
1 |
5 |
14 |
16 |
0,473 |
||||
|
оро-моторные функции |
7 |
13 |
2 |
0 |
5 |
8 |
16 |
7 |
0,007** |
||||
|
апраксия движений рук |
0 |
10 |
7 |
7 |
0 |
0 |
1 |
4 |
9 |
22 |
0, 006** |
||
|
стереотипные движения рук |
0 |
6 |
15 |
3 |
0 |
3 |
19 |
14 |
0,020* |
||||
|
спастичность мышц конечностей |
11 |
11 |
2 |
0 |
13 |
12 |
9 |
2 |
0,043* |
||||
|
патологические рефлексы |
14 |
2 |
6 |
1 |
22 |
10 |
9 |
1 |
0,693 |
||||
|
альтернирующее косоглазие |
9 |
10 |
5 |
0 |
15 |
10 |
13 |
4 |
0,343 |
||||
|
бруксизм |
3 |
9 |
9 |
3 |
1 |
11 |
13 |
11 |
0,041* |
||||
|
сколиоз |
7 |
8 |
8 |
1 |
6 |
11 |
13 |
6 |
0,039* |
||||
|
вертикализация |
9 |
6 |
4 |
4 |
1 |
0 |
6 |
8 |
4 |
11 |
6 |
1 |
0,035* |
|
ходьба |
0 |
1 |
10 |
2 |
2 |
6 |
0 |
1 |
8 |
4 |
2 |
21 |
0,029* |
|
преодоление препятствий |
1 |
6 |
4 |
5 |
1 |
3 |
10 |
22 |
0,064 |
||||
|
судороги |
6 |
9 |
7 |
2 |
10 |
12 |
13 |
1 |
0,789 |
||||
|
тремор головы и туловища |
13 |
6 |
5 |
0 |
12 |
10 |
10 |
4 |
0,110 |
||||
|
вегетативные нарушения |
5 |
11 |
8 |
5 |
11 |
20 |
0,145 |
||||||
|
апноэ и гипервентиляция |
7 |
5 |
10 |
2 |
10 |
9 |
8 |
9 |
0,622 |
||||
|
масса тела |
9 |
2 |
1 |
6 |
11 |
7 |
8 |
10 |
0,379 |
||||
|
длина тела |
14 |
5 |
21 |
0 |
16 |
6 |
8 |
6 |
0,210 |
||||
|
окружность головы |
3 |
3 |
9 |
9 |
3 |
3 |
5 |
25 |
0,027* |
||||
|
длина стопы |
3 |
6 |
2 |
6 |
11 |
5 |
6 |
10 |
0,571 |
||||
Примечания: * р < 0,05; ** р < 0,01.
При использовании разработанной нами клинической шкалы было показано, что фенотип больного с RTT зависит от типа мутации и является более тяжелым при нонсенс мутациях и мутациях со сдвигом рамки считывания по сравнению с миссенс мутациями. При этом можно выделить симптомы, экпрессивность которых наиболее зависит от типа мутации: это нарушения экспрессивной речи, статических (вертикализации) и оро-моторных функций (жевания и глотания), а также целенаправленных движений рук. Вопреки результатам предшествующих работ, зависимости таких дыхательных нарушений, как апноэ и гипервентиляция от типа мутации в наблюдаемой группе детей не обнаружено.
Поскольку различные домены белка MECP2 выполняют различные функции, то фенотипические проявления могут зависеть от позиции мутации в гене MECP2 [Vorsanova et al., 2004]. В нашей работе был проведен анализ экспрессивности заболевания в зависимости от позиции мутации в гене (рис. 39). Обнаружено, что наибольшая тяжесть течения заболевания наблюдается у больных с мутациями, локализованными между 473 и 808 нуклеотидами кодирующей области гена МЕСР2, что соответствует 158–270 аминокислотным остаткам белка. Наиболее тяжелые клинические проявления имели дети с мутациями R168X (локализована между доменами), R255X и R270X (в TRD-домене), наименьшую тяжесть течения болезни – дети
с мутациями R133C (в MBD-домене), мутациями, расположенными ближе к 3′-концу гена – R294X, R306C, и делециями на 3′-конце, что соответствует результатам ряда ранее проведенных зарубежных исследований [Cheadle et al., 2000; Weaving et al., 2003; Neul et al., 2008]. На основании оценки фенотипа по предлагаемой в нашей работе шкале, можно было предположить тип мутации, а иногда и конкретную мутацию у ребенка с RTT. Действительно, у детей с высокой балльной оценкой по шкале – более 45 – выявлялись такие мутации из числа рекуррентных, как R168X, R255X и R270X, а у больных с низкой балльной оценкой – мутации R133C, R294X и R306C.

Рис. 39. Оценка зависимости тяжести течения синдрома Ретта от позиции мутации
Классическая форма заболевания наблюдалась при большинстве обнаруженных мутаций гена MECP2, что было в значительной степени обусловлено строгим клиническим отбором больных для данного исследования. Вариант RTT c сохранной речью установлен у 2-х детей с одинаковой мутацией – R306C. Ранее некоторыми авторами также обращалось внимание на связь этой формы болезни с мутацией R306C, которая нами подтвердилась [Zappella et al., 2001]. Стёртая форма синдрома (forme fruste) отмечалась при мутациях S65X (1 случай), R133C (2 случая), R250fs (1), R294X (1), R306С (1) и делеции 356del92 (1), так же как и в зарубежных исследованиях [RettBASE, IRSF MECP2 Variation Database]. Таким образом, установленные нами у детей с мутациями гена MECP2 клинические формы заболевания совпадали с наблюдавшимися другими авторами при тех же мутациях. Исключение составила классическая форма RTT у мальчика с соматическим мозаицизмом по мутации R168X, которая не выявлялась ранее.
У девочки 4х лет с «RTT-подобным» фенотипом обнаружена миссенс мутация T197M, вызванная заменой цитозина на тимин в положении с.590 и расположенная в четвертом экзоне между MBD и TRD. На основании положения данной миссенс мутации ранее предполагалось, что она не имеет функциональных последствий [Hoffbuhr et al., 2001; Laccone et al., 2002], в то же время другими исследователями была показана связь данной мутации с атипичной формой RTT [Bourdon et al., 2003; Shibayama et al., 2004]. В международной базе данных RettBASE по мутациям гена MECP2 содержатся упоминания о нескольких случаях выявления данной мутации: 2 случая у больных с RTT, 1 – с аутизмом, 1 – с умственной отсталостью. Суммируя литературные данные о мутации T197M, и учитывая то, что она была обнаружена в данной работе у девочки с RTT-подобным фенотипом, можно предположить, что эта мутация является причиной определённого субфенотипа синдрома.
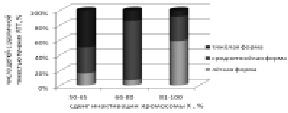
Рис. 40. Зависимость тяжести течения синдрома Ретта от степени сдвига Х-инактивации. Показано уменьшение доли детей с тяжелыми и возрастание больных с лёгкими формами RTT при увеличении сдвига Х-инактивации
Для изучения влияния инактивации хромосомы Х на тяжесть фенотипических проявлений болезни дети были разделены на три группы: с легким течением (10 детей), у которых суммарная оценка фенотипа в баллах составляла менее 30, со средней тяжестью фенотипических проявлений заболевания (19 больных) с суммарной оценкой от 30 до 44 и тяжелым течением болезни (9 больных) с суммарной оценкой более 44 баллов. По степени сдвига инактивации хромосомы Х детей также разделили на три группы: первая – от 50 до 65 (12 детей); вторая – от 66 до 80 (14 детей); третья – более 80 (11 детей). На рис. 40 показана зависимость легкого, среднего и тяжелого течения RTT от различной степени сдвига Х-инактивации. Видно, что среди детей из первой группы с отсутствием сдвига Х-инактивации половина представлена больными с тяжелым течением заболевания. Среди девочек, составляющих вторую группу со сдвигом Х-инактивации 66–80 %, бóльшая часть имеет среднетяжёлую форму синдрома. В третьей группе со сдвигом Х-инактивации более 80 % у основной массы детей наблюдались лёгкие формы болезни. Таким образом, сдвиг инактивации хромосомы Х у детей с RTT в большинстве случаев приводил к более лёгкому течению заболевания, чем у девочек с равной Х-инактивацией, вероятно, из-за преимущественной инактивации хромосомы Х с мутацией. Полученные результаты противоречат тем ранее опубликованным, в которых не было выявлено статистически значимой связи тяжести течения заболевания с типом и позицией мутации [Amir et al., 1999; Huppke et al., 2000; Hoffbuhr et al., 2001], но согласуются с работами, в которых она была показана [Юров и др., 2007; Colvin et al., 2004; Bebbington et al., 2008].
Индивидуальный анализ мутации гена MECP2 и особенностей инактивации хромосомы Х у ребёнка с RTT даёт возможность прогнозировать тяжесть течения заболевания, что наблюдалось в нашем исследовании. Ниже представлены примеры индивидуального анализа корреляции мутации гена MECP2 и особенностей инактивации хромосомы Х с тяжестью клинических проявлений при RTT. Наибольший интерес представлял данный анализ у наблюдавшихся нами близнецов с RTT, фенотип которых представлен на рис. 41.
Краткое клиническое описание случая RTT у близнецов. Родители близнецов не имели родственников с подтвержденной наследственной патологией, брак неродственный. Первая девочка из пары близнецов родилась в головном предлежании с массой тела 1800 г, длиной тела 43 см, окружностью головы 31 см. Оценка по шкале Апгар была 4/7. Первоначально психомоторное развитие девочки было нормальным: уверенно держала голову к 3 мес., хорошо брала предметы и манипулировала ими к 5 мес., сидела с 8 мес., ходила с 16 мес., использовала несколько слов с 8 мес., к году владела 10–12 словами. На втором году жизни развитие приостановилось. В 18 мес. общение и речь стали ухудшаться. К 24–26 мес., утратила целенаправленные движения рук, но сохранила способность ходить. С 2,5 лет появились стереотипии в виде стискивания ладоней. В 3 года – не говорила, выявлялась микроцефалия – окружность головы – 47 см (3й перцентиль). Целенаправленные движения рук были заменены на стереотипные. Отмечались стопы малого размера с вазомоторными нарушениями. Девочка стояла с поддержкой, но не ходила. Были выражены гипотония мышц и умственная отсталость. В 5 лет состояние ребенка ухудшилось: отмечен кифосколиоз, приступы гипервентиляции, пирамидные нарушения, бруксизм. В отличие от сестры-близнеца, она не имела судорог. В целом следует отметить более позднюю манифестацию большинства клинических признаков у (1) по сравнению с (2). Вторая девочка из пары близнецов родилась в тазовом
предлежании с массой тела 1700 г, длиной тела 42 см, с окружностью головы 30 см. Оценка по шкале Апгар была 3/7 баллов. Со слов родителей она начала держать голову к 3 мес., сидела с 9 мес., ходить начала с 18 мес. К году говорила 4–5 слов. Девочка хорошо удерживала и манипулировала предметами, активно общалась и играла. В возрасте 12 мес. перестала произносить новые слова, а в 17мес., после бронхита ухудшился контакт ребенка с окружающими, стал очевиден регресс речевых навыков. Однако девочка стала самостоятельно, но неустойчиво ходить. Родители сообщают об утрате целенаправленных движений рук, нарушении сна и приступах немотивированного крика в возрасте 20 месяцев. К 2 г 2 мес. она утратила навык ходьбы. В это же время появились стереотипные движения в виде стискивания ладоней и стучания дорсальной поверхностью кистей по зубам. При обследовании в трехлетнем возрасте больная имела хороший эмоциональный контакт с родителями, развитие ее было на уровне ребенка в возрасте 10 мес. Выявлялась микроцефалия – окружность головы – 47 см (менее 3х перцентилей), выраженная диффузная гипотония мышц. Обращал на себя внимание малый размер стоп, а также вазомоторные расстройства в дистальных отделах ног. Целенаправленные движения рук отсутствовали, наблюдались стереотипии, периодически возникало сходящееся косоглазие. Несколько раз в неделю отмечались абсансы и кратковременные фокальные судороги. При осмотре в возрасте 5 лет отмечен кифосколиоз, пирамидные расстройства, пароксизмы гипервентиляции. В этом возрасте близнецы были помещены в интернат для детей с умственной отсталостью, где вторая девочка умерла в 8-летнем возрасте во время эпилептического приступа.

Рис. 41. Фенотип близнецов с синдромом Ретта и мутацией R255X гена MECP2
У двух близнецов с классической формой RTT определена одинаковая нонсенс мутация R255X, тогда как у их матери данная мутация не была обнаружена. Анализ инактивации хромосомы Х у первой девочки выявил значительный сдвиг Х-инактивации (80:20), у второй – равную Х-инактивацию (56:44), у матери Х-инактивация составила 77:23. Сдвиг Х-инактивации у матери не является значительным, поэтому носительство ею МЕСР2 мутации было исключено. Обнаружена также одинаковая длина полиморфного участка ЦАГ-повторов в гене андрогенного рецептора (AR) у обеих девочек, что вместе с наличием одинаковой МЕСР2 мутации дало основание полагать, что эти близнецы являются монозиготными. Мутация R255X гена МЕСР2 приводит к потере основных функций белка МEСР2 и в большинстве случаев – к очень тяжелому течению синдрома [Bebbington et al., 2008]. Однако нами наблюдалось более легкое течение заболевания у девочки со сдвигом Х-инактивации, чем у девочки с равной инактивацией хромосомы Х. Об этом свидетельствует то, что количественная оценка клинических проявлений RTT была 49 баллов у первой и 57 баллов у второй девочки. Регресс, появление стереотипий и утрата целенаправленных движений рук у девочки с неравной Х-инактивацией появились позже, чем у сестры, а также у неё не наблюдалось судорог, тогда как у девочки с равной Х-инактивацией судорожный синдром имел крайне тяжелое течение, что привело к её смерти в возрасте 8 лет (табл. 24).
Таблица 24
Сравнительной анализ тяжести течения RTT у монозиготных близнецов с мутацией R255X
|
Тяжесть течения и возраст появления признаков |
Близнецы |
|
|
Больная 1 |
Больная 2 |
|
|
Возраст начала регресса, мес. |
18 |
17 |
|
Сумма баллов количественной оценки фенотипа |
49 |
57 |
|
Возраст появления судорожных пароксизмов, мес. |
– |
36 |
|
Появление стереотипий, мес. |
30 |
26 |
|
Утрата целенаправленных движений рук, мес. |
24–26 |
20 |
|
Летальный исход |
– |
В возрасте 8 лет |
Результаты обследования данной семьи свидетельствуют о том, что при преимущественной инактивации мутантной хромосомы Х может наблюдаться более легкое течение болезни. В то же время нами наблюдалась еще одна семья, в которой близнецы страдали RTT и имели одну и ту же мутацию – R270X. У этих девочек практически не было различий в тяжести течения болезни и сроках её манифестации (рис. 42). У обеих девочек стадия регресса началась в 1 г 2 месяца, в возрасте 4 года суммарная оценка фенотипа составила 44 и 45 баллов, а Х-инактивация носила случайный характер.


Рис. 42. Фенотип близнецов с синдромом Ретта и мутацией R270X: слева девочки в возрасте 13 месяцев перед началом стадии регресса; справа – близнецы в возрасте 4х лет во время псевдостационарной стадии болезни
Особенности Х-инактивации имеют значительное влияние на формирование фенотипических признаков при RTT. При сдвиге Х-инактивации против хромосомы Х без мутации будет наблюдаться более тяжелое течение болезни. Так, у одной из больных с классической формой синдрома обнаружена мутация R168X и сдвиг Х-инактивации 90:10 против материнской хромосомы Х. Анализ Х-инактивации у матери девочки показал её случайный характер. В данном случае, по-видимому, инактивирована немутантная хромосома Х, что объясняет тяжелую форму болезни у ребенка (суммарный показатель оценки тяжести фенотипа – 46 баллов). Суммируя полученные данные, следует заключить, что существуют корреляции типа и позиции мутации гена МЕСР2, а также особенности инактивации хромосомы Х с тяжестью течения RTT и экспрессивностью его отдельных признаков. Полученные корреляции генотип/фенотип помогают понять основы клинического полиморфизма RTT и предположить тяжесть течения болезни ребенка уже в раннем возрасте (табл. 25). Система прогнозирования тяжести состояния ребёнка с RTT должна основываться на исследовании мутаций гена MECP2 и знании о влиянии конкретных мутаций на тяжесть фенотипических проявлений, а также на определении особенностей инактивации хромосомы Х. При этом следует учитывать то, что сдвиг Х-инактивации против мутантной хромосомы Х может облегчать течение болезни, а сдвиг Х-инактивации против нормальной хромосомы Х может делать его более тяжёлым.
Система прогнозирования тяжести течения синдрома FRAXA также предложена нами. Анализ зависимости фенотипа индивидуумов из семей с синдромом FRAXA от наличия мутации/премутации гена FMR1 и особенностей инактивации хромосомы Х проведен в 27 семьях с данной патологией. Для этого сравнивались фенотипы двух групп детей:
1) мальчиков, у которых определена мутация гена FMR1 (27 пациентов),
2) мальчиков с недифференцированной умственной отсталостью, аутизмом и фенотипическими признаками, напоминающими синдром FRAXA, у которых определено нормальное число триплетных повторов в промоторе гена FMR1 (41 пациент).
Таблица 25
Прогнозирование тяжести течения синдрома Ретта на основе определения мутации гена MECP2 и анализа инактивации хромосомы Х
|
Генетические и эпигенетические факторы, определяющие тяжесть течения RTT |
Тяжёлое течение |
Легкое течение |
|
Тип и позиция мутации |
● Нонсенс мутации ● Мутации со сдвигом рамки считывания за исключением делеций на 3’-конце гена MECP2 |
● Миссенс мутации ● Делеции на 3’-конце гена MECP2 |
|
Рекуррентные мутации |
● R168X ● R255X ● R270X ● T158M |
● R106W ● R133C ● R294X ● R306C |
|
Особенности инактивации хромосомы Х |
● Равная инактивация хромосомы Х ● Преимущественная инактивация хромосомы Х с нормальным аллелем гена MECP2 |
● Преимущественная инактивация хромосомы Х с мутацией гена MECP2 |
Дети были обследованы с помощью ранговой шкалы, разработанной для количественной оценки степени тяжести клинических симптомов заболевания. Результаты суммарной оценки фенотипа детей по клинической шкале, данные о регрессе психического развития, случаях ранней менопаузы и синдрома тремора и атаксии у родственников I–II степеней родства представлены в табл. 26.
Таблица 26
Результаты клинического обследования больных с синдромом FRAXA и детей с недифференцированными формами умственной отсталости и аутизма, а также их родственников
|
№ индивидуума |
Возраст, лет |
Суммарная оценка по клинической шкале, баллы |
Регресс психического развития в анамнезе |
Кол-во случаев преждевременного нарушения функции яичников у родственников I–II степеней родства |
Кол-во случаев синдрома тремора и атаксии у родственников I–II степеней родства |
|
Дети с синдромом FRAXA, у которых определена полная мутация гена FMR1 |
|||||
|
1 |
7 |
49 |
– |
1 |
1 |
|
2 |
7 |
39 |
– |
0 |
0 |
|
3 |
10 |
38 |
– |
0 |
0 |
|
4 |
16 |
40 |
– |
1 |
0 |
|
5 |
11 |
32 |
– |
0 |
1 |
|
6 |
15 |
37 |
– |
0 |
0 |
|
7 |
14 |
36 |
– |
0 |
0 |
|
8 |
5 |
36 |
– |
0 |
0 |
|
9 |
15 |
24 |
– |
0 |
1 |
|
10 |
17 |
42 |
– |
1 |
0 |
|
11 |
12 |
37 |
– |
1 |
0 |
|
12 |
15 |
47 |
– |
0 |
1 |
|
13 |
17 |
36 |
– |
0 |
0 |
|
14 |
15 |
42 |
– |
1 |
0 |
|
15 |
10 |
45 |
– |
0 |
0 |
|
16 |
8 |
40 |
– |
0 |
1 |
|
17 |
11 |
43 |
– |
0 |
0 |
|
18 |
6 |
27 |
– |
0 |
0 |
|
19 |
8 |
40 |
– |
0 |
0 |
|
20 |
7,5 |
33 |
– |
1 |
1 |
|
21 |
14,5 |
39 |
– |
0 |
0 |
|
22 |
4,5 |
39 |
– |
0 |
0 |
|
23 |
9 |
41 |
– |
2 |
1 |
|
24 |
13 |
39 |
– |
0 |
0 |
|
25 |
6 |
47 |
– |
0 |
0 |
|
26 |
15 |
42 |
– |
1 |
0 |
|
27 |
9 |
37 |
– |
0 |
0 |
|
Дети с недифференцированными формами умственной отсталости и аутизма, у которых определено нормальное число CGG-повторов в гене FMR1 |
|||||
|
28 |
6 |
20,5 |
– |
0 |
0 |
|
29 |
6 |
21 |
– |
0 |
0 |
|
30 |
8 |
22 |
– |
0 |
0 |
|
31 |
5 |
15 |
– |
0 |
0 |
|
32 |
10 |
29,5 |
+ |
0 |
0 |
|
33 |
7 |
18 |
– |
0 |
0 |
|
34 |
9 |
25,5 |
+ |
0 |
0 |
|
35 |
5 |
28 |
– |
0 |
0 |
|
36 |
5 |
14 |
– |
0 |
0 |
|
37 |
8,5 |
21 |
– |
0 |
0 |
|
38 |
12 |
21,5 |
– |
0 |
0 |
|
39 |
4,5 |
20 |
– |
0 |
0 |
|
40 |
4 |
16 |
– |
0 |
0 |
|
41 |
9 |
27,5 |
+ |
0 |
0 |
|
42 |
10 |
24 |
– |
0 |
0 |
|
43 |
15 |
30,5 |
+ |
0 |
0 |
|
44 |
6 |
29 |
– |
0 |
0 |
|
45 |
13 |
33 |
– |
0 |
0 |
|
46 |
11 |
17 |
– |
0 |
0 |
|
47 |
7 |
34,5 |
+ |
0 |
0 |
|
48 |
16 |
15 |
– |
0 |
0 |
|
49 |
12 |
27 |
– |
0 |
0 |
|
50 |
6 |
17 |
– |
0 |
0 |
|
51 |
11 |
32 |
+ |
0 |
0 |
|
52 |
14 |
31 |
– |
0 |
0 |
|
53 |
5 |
16 |
– |
0 |
0 |
|
54 |
11 |
29 |
+ |
0 |
0 |
|
55 |
7 |
19 |
– |
0 |
0 |
|
56 |
16 |
26 |
– |
0 |
0 |
|
57 |
10 |
30 |
+ |
0 |
0 |
|
58 |
6 |
18 |
– |
0 |
0 |
|
59 |
11,5 |
24,5 |
– |
0 |
0 |
|
60 |
15 |
22 |
– |
0 |
0 |
|
61 |
12 |
29 |
– |
0 |
0 |
|
62 |
5 |
27,5 |
– |
0 |
0 |
|
63 |
8 |
31 |
+ |
0 |
0 |
|
64 |
13 |
34 |
+ |
0 |
0 |
|
65 |
10 |
16 |
– |
0 |
0 |
|
66 |
7,5 |
19 |
– |
0 |
0 |
|
67 |
12 |
28 |
+ |
0 |
0 |
|
68 |
13 |
26,5 |
– |
0 |
0 |
Анализ данных, представленных в табл. 26, показал, что суммарная балльная оценка по клинической шкале у детей с синдромом FRAXA была статистически достоверно выше, чем у больных с недифферецированными формами умственной отсталости (критерий Манна-Уитни, p < 0,001). Кроме того, в анамнезе у детей с синдромом FRAXA, в отличие от больных с недифференцированными формами умственной отсталости и аутизма, не встречалось регресса психического развития, а случаи преждевременного нарушения функции яичников у женщин наблюдались только в родословных детей с синдромом FRAXA. Для того чтобы определить, какие признаки (особенности фенотипа, течения заболевания) отличают больных с мутацией от детей без таковой
проведено сравнение экспрессивности клинических симптомов у данных групп индивидуумов с помощью непараметрического теста Манна-Уитни для двух независимых выборок (табл. 27). Определены статистически значимые различия между двумя группами детей по тяжести следующих признаков: зрительный и тактильный контакт, тахилалия (ускоренный темп речи), речевые персеверации и эхолалия, специфические стереотипные движения в виде «встряхивания кистей», комплекс лицевых микроаномалий (удлиненная форма лица, загнутый вниз кончик носа, крупные оттопыренные ушные раковины, выступающий лоб и подбородок), макроорхизм, аномалии соединительной ткани (гиперрастяжимость кожи, гиперподвижность суставов, плоскостопие) и увеличение окружности головы. Перечисленные признаки были в большей степени выражены у детей с синдромом FRAXA. Признаки, наблюдавшиеся у женщин-родственниц пробанда с мутациями и премутациями гена FMR1, представлены в табл. 28.
Таблица 27
Сравнение экспрессивности клинических признаков, выраженной в баллах, у детей с синдромом FRAXA (N = 27) и детей с недифференцированной умственной отсталостью и аутизмом (N = 41) с использованием непараметрического критерия Манна-Уитни
|
Признаки |
Количество детей, получивших оценку в баллах: |
p |
|||||||||||
|
синдром FRAXA (N = 27) |
недифференцированная умственная отсталость |
||||||||||||
|
0 |
1 |
2 |
3 |
4 |
5 |
0 |
1 |
2 |
3 |
4 |
5 |
||
|
контакт |
6 |
19 |
5 |
0 |
10 |
13 |
8 |
0 |
0,303 |
||||
|
зрительная реакция |
4 |
9 |
14 |
3 |
9 |
13 |
19 |
0 |
0,032* |
||||
|
тактильный контакт |
3 |
18 |
9 |
16 |
23 |
2 |
0,014* |
||||||
|
невербальное общение |
1 |
18 |
8 |
3 |
3 |
22 |
16 |
0 |
0,561 |
||||
|
экспрессивная речь |
0 |
17 |
8 |
3 |
1 |
1 |
8 |
16 |
14 |
3 |
0 |
0,470 |
|
|
умение использовать |
5 |
12 |
7 |
1 |
14 |
14 |
7 |
6 |
0,375 |
||||
|
ускоренный темп речи |
5 |
9 |
11 |
27 |
6 |
3 |
< 0,001*** |
||||||
|
аграмматизмы, речевые персеверации и эхолалия (исключены дети без речи) |
1 |
2 |
8 |
14 |
14 |
22 |
0 |
0 |
0,008** |
||||
|
гипотония мышц |
2 |
21 |
6 |
1 |
11 |
27 |
3 |
0 |
0,056 |
||||
|
комплекс |
0 |
1 |
2 |
3 |
9 |
15 |
0 |
3 |
17 |
14 |
5 |
2 |
< 0,001*** |
|
макроорхизм |
3 |
6 |
18 |
25 |
12 |
4 |
< 0,001*** |
||||||
|
гиперрастяжимость кожи |
4 |
20 |
6 |
17 |
21 |
3 |
0,009** |
||||||
|
гиперподвижность |
3 |
3 |
5 |
8 |
11 |
9 |
8 |
6 |
7 |
11 |
0,425 |
||
|
плоскостопие |
6 |
12 |
12 |
13 |
18 |
10 |
0,031* |
||||||
|
нарушения осанки |
11 |
13 |
6 |
28 |
10 |
3 |
0,006** |
||||||
|
длина тела |
15 |
5 |
1 |
9 |
27 |
9 |
3 |
2 |
0,059 |
||||
|
окружность головы |
16 |
10 |
2 |
2 |
31 |
8 |
2 |
0 |
0,024* |
||||
Примечания: *p < 0,05; ** p < 0,01; *** p < 0,001.
Таблица 28
Клинические признаки, количество триплетных повторов в промоторе гена FMR1
и особенности инактивации хромосомы Х у родственниц пробандов с синдромом FRAXA
|
№ семьи |
Родство по отношению к пробанду |
Возраст, лет |
Патологические признаки |
Число CGG-повторов в гене FMR1 |
Х-инактивация |
% клеток с активной хромосомой Х, несущей аллель гена FMR1 c экспансией CGG-повторов |
|
1 |
Мать |
36 |
Нет нарушений |
125 |
99:1 |
1 |
|
Сестра |
5 |
Нет нарушений |
> 200 |
5:95 |
5 |
|
|
2 |
Мать |
38 |
Нет нарушений |
88 |
18: 82 |
18 |
|
Сестра |
14 |
Легкая умственная отсталость, гиперподвижность суставов и повышенная растяжимость кожи, удлиненное лицо, большие оттопыренные ушные раковины |
> 200 |
80:20 |
80 |
|
|
3 |
Сестра |
32 |
Нарушение обучения |
> 200 |
63:37 |
37 |
|
4 |
Сестра |
40 |
Нарушение памяти |
> 200 |
35:65 |
35 |
|
5 |
Двоюродная сестра по линии матери |
6 |
Умственная отсталость, небольшая гиперподвижнсть суставов, большие оттопыренные ушные раковины, выступающий лоб |
> 200 |
39:61 |
61 |
|
Тетя по линии матери |
28 |
Нет нарушений |
90 |
75:25 |
25 |
|
|
Мать |
33 |
Нарушение обучения, дисфункция яичников |
91 |
61:39 |
39 |
|
|
6 |
Бабушка по линии матери |
66 |
Менопауза с 35 лет |
70 |
23:77 |
23 |
|
Мать |
39 |
Дисфункция яичников с 30 летнего возраста |
101 |
60:40 |
40 |
|
|
7 |
Мать |
- |
Умерла в 42 года от рака шейки матки, в детстве выраженные нарушения в обучении |
– |
– |
– |
|
Бабушка по линии матери |
70 |
Оперирована по поводу опухоли яичников |
72 |
18:82 |
18 |
|
|
8 |
Мать |
26 |
Нарушение памяти и внимания, тревожность |
62 |
68:32 |
68 |
|
9 |
Мать |
45 |
Оперирована по поводу миомы матки |
75 |
19:81 |
19 |
|
10 |
Мать |
40 |
Крупные ушные раковины |
119 |
19:81 |
19 |
|
11 |
Мать |
52 |
Менопауза с 38 лет |
79 |
31:69 |
31 |
|
12 |
Мать |
30 |
Дисфункция яичников |
117 |
21:79 |
21 |
|
13 |
Мать |
29 |
Нарушения обучения |
80 |
31:69 |
31 |
|
14 |
Мать |
30 |
Нет нарушений |
89 |
8:92 |
8 |
Когнитивные нарушения выявлялись у родственниц-гетерозигот по полной мутации гена FMR1 (у родных и двоюродных сестер). Нарушения интеллекта у индивидуумов женского пола имели более лёгкую степень выраженности, чем у мальчиков с мутацией данного гена: обнаружены нарушения памяти и обучения, лёгкая и умеренная умственная отсталость, задержка психомоторного развития. У этих сестер когнитивные нарушения сочетались с микроаномалиями и соединительно-тканными нарушениями, характерными для синдрома FRAXA, что дало основание предположить у них диагноз данного заболевания. Нами обследованы 23 женщины с премутацией гена FMR1. Отсутствие клинических признаков наблюдалось у восьми из 23-х женщин. Пять женщин имели различные когнитивные расстройства: снижение памяти (2 пациентки), способности к обучению (2 пациентки) и пограничный с умственной отсталостью интеллект (1 пациентка), у 3-х из них когнитивные нарушения сочетались с аномалиями поведения: тревожными расстройствами (2 пациентки) и отсутствием чувства дистанции в общении (1 пациентка). У одной из матерей помимо нарушений интеллекта и поведения периодически наблюдался тремор головы, что указывало на развитие у неё синдрома тремора и атаксии, характерного для индивидуумов с премутацией гена FMR1. Для гетерозигот по премутации гена FMR1 были характерны гинекологические заболевания, которые не встречались у женщин с полными мутациями. Степень выраженности когнитивных нарушений была выше при полной мутации данного гена. Микроаномалии, характерные для синдрома FRAXA, выявлялись как при полной мутации, так и при премутации гена FMR1. Следует упомянуть также о единственном наблюдавшемся нами мужчине с премутацией гена FMR1 (дед пробанда по линии матери из семьи 14), у которого в возрасте 56 лет наблюдались выраженные признаки синдрома тремора и атаксии (FXTAS). Связь раннего наступления менопаузы и развития синдрома тремора и атаксии с премутацией гена FMR1 подчёркивалась ранее многими авторами [Bodega et al., 2006; Bourgeois et al., 2009]. Отмеченное нами наличие опухолей репродуктивных органов у женщин с премутацией было определено в единичных зарубежных исследованиях и требует дальнейшего накопления фактов [Allen et al., 2007]. Полученные данные об отсутствии гинекологической патологии у носительниц полной мутации подтверждают результаты, опубликованные ранее другими исследователями [Sherman et al., 2005].
Исследования инактивации хромосомы Х проведены у 19 индивидуумов женского пола – гетерозигот по мутациям и премутациям
гена FMR1 из семей мальчиков с синдромом FRAXA. Анализ результатов показал, что чем выше был процент клеток с активной хромосомой Х с полной мутацией, тем более выражены были когнитивные нарушения у женщин. Похожие на наши данные о влиянии особенностей инактивации хромосомы Х на тяжесть нарушений интеллекта у гетерозигот с мутацией гена FMR1 получены ранее другими исследователями [De Vries et al.,1997; Berry-Kravis et al., 2005]. Среди женщин с премутацией корреляций между процентом клеток с активной хромосомой Х с премутацией и клиническими проявлениями проследить не удалось. Это могло быть связано с тем, что особенности Х-инактивации в клетках крови и других органов могут не совпадать. Кроме того, возраст более половины женщин был менее 40 лет, когда могут еще отсутствовать признаки преждевременного нарушения функции яичников или синдрома тремора и атаксии, связанных с премутацией гена FMR1, так как эти заболевания проявляются в более позднем возрасте. Однако отдельными авторами, наблюдавшими женщин – носительниц премутации гена FMR1 в возрасте старше 40 лет, также обсуждалось отсутствие связи между величиной и направлением сдвига Х-инактивации и риском раннего наступления менопаузы [Spath et al., 2010]. Поскольку в работе выявлена зависимость фенотипа индивидуумов от наличия мутации/премутации гена FMR1 и особенностей инактивации хромосомы Х, то эти данные можно использовать для прогнозирования течения болезни (табл. 29).
Таблица 29
Фенотип на основе определения числа CGG-повторов в гене FMR1 и анализа инактивации хромосомы Х
|
Генетические и эпигенетические факторы, определяющие фенотип |
Фенотип индивидуумов мужского пола |
Фенотип индивидуумов |
|
Число CGG-повторов в гене FMR1 от 55 до 200 |
– Когнитивные нарушения и нарушения поведения – Повышен риск развития синдрома FXTAS (тремора и атаксии) |
Когнитивные нарушения и нарушения поведения. Повышен риск развития: – дисфункции яичников, – синдрома POF (менопауза до 40-летнего возраста), – опухолей женских половых органов, – синдрома FXTAS |
|
Число CGG-повторов в гене FMR1 более 200 |
– Синдром умственной отсталости, сцепленной с ломкой хромосомой Х |
– Синдром умственной отсталости, сцепленной с ломкой хромосомой Х – Асимптоматическое носительство мутации |
|
Особенности инактивации хромосомы Х |
– |
Снижение интеллекта у женщин с полной мутацией гена FMR1 пропорционально проценту клеток с активной мутантной хромосомой Х |
Необходимо учитывать, что полная мутация у мальчиков-гемизигот ведёт к клиническим проявлениям синдрома FRAXA, в то время как у гетерозигот по полной мутации гена FMR1 на тяжесть клинических проявлений значительное влияние оказывают особенности инактивации хромосомы Х. При наличии премутации гена FMR1 существует риск возникновения с возрастом синдрома преждевременного прекращения активности яичников и синдрома тремора и атаксии. У индивидуумов с премутацией следует ожидать развития когнитивных расстройств и нарушений поведения лёгкой степени, а также дисфункции яичников и новообразований женских половых органов в молодом возрасте. Носители премутаций должны составлять группу риска по указанным нарушениям, в которой рекомендуется проводить профилактические мероприятия.